紫外光谱
紫外光谱,准确测定有机化合物的分子结构,对从分子水平去认识物质世界,推动近代有机化学的发展是十分重要的。采用现代仪器分析方法,可以快速、准确地测定有机化合物的分子结构。在有机化学中应用最广泛的测定分子结构的方法是四大光谱法:紫外光谱、红外光谱、核磁共振和质谱。紫外和可见光谱(ultraviolet and visible spectrum)简写为UV。[1]
| 紫外光谱 | |
|---|---|

{kind=link}
目录
基本原理
光谱的产生
在紫外光谱中,波长单位用nm(纳米)表示。紫外光的波长范围是10~380 nm,它分为两个区段。波长在10~200 nm称为远紫外区,这种波长能够被空气中的氮、氧、二氧化碳和水所吸收,因此只能在真空中进行研究工作,故这个区域的吸收光谱称真空紫外,由于技术要求很高,目前在有机化学中用途不大。波长在200~380 nm称为近紫外区,一般的紫外光谱是指这一区域的吸收光谱。波长在400~750 nm范围的称为可见光谱。常用的分光光度计一般包括紫外及可见两部分,波长在200~800 nm(或200~1000 nm)。
分子内部的运动有转动、振动和电子运动,相应状态的能量(状态的本征值)是量子化的,因此分子具有转动能级、振动能级和电子能级。通常,分子处于低能量的基态,从外界吸收能量后,能引起分子能级的跃迁。电子能级的跃迁所需能量最大,大致在1~20 eV(电子伏特)之间。根据量子理论,相邻能级间的能量差ΔE、电磁辐射的频率ν、波长λ符合下面的关系式
ΔE=hν=h×c/λ
式中h是普朗克常量,为6.624×10⁻³⁴J·s=4.136×10⁻¹⁵ eV·s;c是光速,为2. 998×10¹⁰ cm/s。应用该公式可以计算出电子跃迁时吸收光的波长。
许多有机分子中的价电子跃迁,须吸收波长在200~1000 nm范围内的光,恰好落在紫外-可见光区域。因此,紫外吸收光谱是由于分子中价电子的跃迁而产生的,也可以称它为电子光谱。
跃迁类型
有机化合物分子中主要有三种电子:形成单键的σ电子、形成双键的π电子、未成键的孤对电子,也称n电子。基态时σ电子和π电子分别处在σ成键轨道和π成键轨道上,n电子处于非键轨道上。仅从能量的角度看,处于低能态的电子吸收合适的能量后,都可以跃迁到任一个较高能级的反键轨道上。跃迁的情况如下图所示:
上图中虚线下的数字是跃迁时吸收能量的大小顺序,该顺序也可以表示为
n→π*<π→π*<n→σ*<π→σ*<σ→π*<σ→σ*
即n→π*的跃迁吸收能量最小。实际上,对于一个非共轭体系来讲,所有这些可能的跃迁中,只有n→π*的跃迁的能量足够小,相应的吸收光波长在200~800 nm范围内,即落在近紫外-可见光区。其它的跃迁能量都太大,它们的吸收光波长均在200 nm以下,无法观察到紫外光谱。但对于共轭体系的跃迁,它们的吸收光可以落在近紫外区。
根据上图,可以认为:烷烃只有σ键,只能发生σ→σ*的跃迁。含有重键如C=C,C≡C,C=O,C=N等的化合物有σ键和π键,有可能发生σ→σ*,σ→π*,π→π*,π→σ*的跃迁。分子中含有氧、卤素等原子时,因为它们含有n电子,还可能发生n→π*、n→σ*的跃迁。
一个允许的跃迁不仅要考虑能量的因素,还要符合动量守恒(跃迁过程中光量子的能量不转变成振动的动能)、自旋动量守恒(电子在跃迁过程中不发生自旋翻转),此外,还要受轨道对称件的制约。即使是允许的跃迁,它们的跃迁概率也是不相等的。有机分子最常见的跃迁是σ→σ*,π→π*,n→σ*,n→π*的跃迁。
电子的跃迁可以分成三种类型:基态成键轨道上的电子跃迁到激发态的反键轨道称为N→V跃迁,如σ→σ*,π→π*的跃迁。杂原子的孤对电子向反键轨道的跃迁称为N→Q跃迁,如n→σ*,n→π*的跃迁。还有一种N→R跃迁,这是σ键电子逐步激发到各个高能级轨道上,最后变成分子离子的跃迁,发生在高真空紫外的远端。
光谱图
紫外光谱图提供两个重要的数据:吸收峰的位置和吸收光谱的吸收强度。从图中可以看出,化合物对电磁辐射的吸收性质是通过一条吸收曲线来描述的。图中以波长(单位nm)为横坐标,它指示了吸收峰的位置在260 nm处。纵坐标指示了该吸收峰的吸收强度,吸光度为0.8。
吸收光谱的吸收强度是用Lambert(朗伯)-Beer(比尔)定律来描述的,这个定律可以用下面的公式来表示:
A=lg(I0/I)=kcl=lg(1/T)
式中A称为吸光度(absorbance)。I0是入射光的强度,I是透过光的强度,T=I/I0为透射比(transmiπance),又称为透光率或透过率,用百分数表示。l是光在溶液中经过的距离(一般为吸收池的长度)。c是吸收溶液的浓度。κ=A/(cl),称为吸收系数(absorptivity)。若c以mol/L为单位,l以cm为单位,则κ称为摩尔消光系数或摩尔吸收系数,单位为c㎡·mol(通常可省略)。
A,T,(1-T)(吸收率),κ,lgκ都能作为紫外光谱图的纵坐标,但最常用的是κ,lgκ。上图是以吸光度A为纵坐标的紫外光谱图,下面四幅图是以T,1-T,κ,lgκ为纵坐标的紫外光谱图。由图可知,透过率与吸收率正好相反,如吸收率为20%,透过率恰好为80%。[2] 最大吸收时的波长(λmax)为紫外的吸收峰,在以吸光度、κ,lgκ、吸收率为纵坐标的谱图中,λmax处于吸收曲线的最高峰顶,而在以透过率为纵坐标的谱图中,λmax处于曲线的最低点。紫外吸收的强度通常都用最大吸收峰的κ值即κmax来衡量。在多数文献报告中,并不绘制出紫外光谱图,只是报道化合物最大吸收峰的波长及与之相应的摩尔消光系数。例如CH₃I的紫外吸收数据为λmax 258 nm(365),这表示吸收峰的波长为258 nm,相应的摩尔消光系数为365。
紫外光谱的测定大都是在溶液中进行的,绘制出的吸收带大都是宽带,这是 因为分子振动能级的能级差为0.05~1 eV,转动能级的能差小于0.05 eV,都远远低于电子能级的能差,因此当电子能级改变时,振动能级和转动能级也不可避免地会有变化,即电子光谱中不但包括电子跃迁产生的谱线,也有振动谱线和转动谱线,分辨率不高的仪器测出的谱图,由于各种谱线密集在一起,往往只看到一个较宽的吸收带。若紫外光谱在惰性溶剂的稀溶液或气态中测定,则图谱的吸收峰上因振动吸收而会表现出锯齿状精细结构。降低温度可以减少振动和转动对吸收带的贡献, 因此有时降温可以使吸收带呈现某种单峰式的电子跃迁。溶剂的极性对吸收带的形状也有影响,通常的规律是溶剂从非极性变到极性时,精细结构逐渐消失,图谱趋向平滑。
电子跃迁
饱和有机化合物
饱和烃分子是只有C-C键与C-H键的分子,只能发生σ→σ*跃迁,由于σ电子不易激发,故跃迁需要的能量较大,即必须在波长较短的辐射照射下才能发生。如CH4的σ→σ*跃迁在125 nm,乙烷的σ→σ*跃迁在135 nm,其它饱和烃的吸收一般波长在150 nm左右,均在远紫外区。
如果饱和烃中的氢被氧、氮、卤素等原子或基团取代,这些原子中的n轨道的电子可以发生n→σ*跃迁。见下图。
n→σ*跃迁 n→σ*跃迁 下表列举了一些能进行跃迁的化合物。
一些化合物发生n→σ*跃迁时的吸收光 化合物
CH₃Cl
CH₃OH
CH₃OCH₃
CH₃Br
CH₃NH₂
CH₃I
λmax
172(弱)
183(150)
185(2520)
204(200)
215(600)
258(365)
从上表可以看出,C-O(醇、醚),C-Cl等基团的n→σ*跃迁,吸收光的波长小于200 nm,在真空紫外,而C一Br,C一I,C-NH₂等基团的n→σ*跃迁,吸收光的波长大于200 nm,可以在近紫外区看到不强的吸收。这些化合物在吸收光谱上的差别,主要是由于原子的电负性不同,原子的电负性强,对电子控制牢,激发电子需要的能量大,吸收光的波长短;反之,原子的电负性较弱,对电子控制不牢,激发电子需要的能量较小,可以在近紫外区出现吸收。此外,分子的可极化性对其吸收光的波长也有一定的影响。可极化性大的,吸收光的波长也较长,n→σ*跃迁的κ值一般在几百以下。
由于饱和烃、醇、醚等在近紫外区不产生吸收,一般用紫外-可见分光光度计无法测出,因此在紫外光谱中常用作溶剂。
不饱和脂肪族化合物
1.π→π*跃迁
C=C双键可以发生π→π*跃迁,由于原子核对π电子的控制不如对σ电子牢,跃迁所需的能量较σ电子小。所以→π*跃迁κ值较大,在5000~100000左右,但是只有一个C=C双键的跃迁出现在170~200 nm处,在真空紫外吸收,一般的分光光度计不能观察到。例如乙烯的π→π*跃迁,λmax= 185 nm(κ=10000),在近紫外区不能检出,同样C≡C与C≡N等π→π*跃迁的吸收亦小于200 nm。
如果分子中存在两个或两个以上的双键(包括三键)形成的共轭体系,π电子处在离域的分子轨道上,与定域轨道相比,占有电子的成键轨道的最高能级与未占有电子的反键轨道的最低能级的能差减小,使π→π*跃迁所需的能量减少,因此吸收向长波方向位移。消光系数也随之增大,例如1,3-丁二烯分子中两对π电子填满π1与π2成键轨道,π3与T4反键轨道是空的,当电子吸收了所需的光能后便会发生从π2到π3的跃迁,见下图。
由图可知,在这种分子中,电子可以有多种跃迁,但是在有机分子中比较重要的是能量最低的跃迁,因为这种跃迁在近紫外区吸收,1,3-丁二烯的能量最低跃迁是π2→π3跃迁,其λmax=217 nm(κ= 21000),而其它跃迁能阶相差较高,需要能量较大,在真空紫外吸收。随着共轭体系逐渐增长,跃迁能阶的能差逐渐变小,吸收愈向长波方向位移,由近紫外可以转向可见光吸收(见下表)。
多烯化合物的吸收带 化合物
双键
λmax/nm(κ)
颜色
乙烯
1
185(10,000)
无色
丁二烯
2
217(21,000)
无色
1,3,5-己三烯
3
285(35,000)
无色
癸五烯
5
335(118,000)
淡黄
二氢-β-胡萝卜素
8
415(210,000)
橙黄
番茄红素
11
470(185,000)
红
因为共扼体系吸收带的波长在近紫外,因此在紫外光谱的应用上,占有重要地位,对于判断分子的结构,非常有用。
2.n→π*跃迁
有些基团存在双键和孤电子对,如C=O,N=O,C=S,N=N等,这些基团除了可以进行π→π*跃迁,有较强的吸收外,还可进行n→π*跃迁,这种跃迁所需能量较少,可以在近紫外或可见光区有不太强的吸收,κ值一般在十到几百。例如脂肪醛中C=O的π→π*跃迁吸收约210 nm,n→π*跃迁吸收约290 nm,见下左图。
如果这些基团与C=C共轭,形成含有杂原子的共轭体系,与C=C-C=C共轭类似,可以形成新的成键轨道与反键轨道,使与π→π*与n→π*的跃迁能级的能差减小,吸收向长波方向位移,例如2-丁烯醛的π2→π3和n→π3跃迁与脂肪醛相应的跃迁比较,吸收均向长波位移,见下右图。
下表列举了常见的n→π*跃迁化合物的吸收带以及不同类型共轭分子的吸收带。
-些化合物的n→π*π→π*跃迁的吸收带 化合物
基团
π→π*λmax/nm(κ)
n→π*λmax/nm(κ)
醛
-CHO
~210(强)
285~295(10~30)
酮
羰基
~195(1000)
270~285()
硫酮
~200(强)
~400(弱)
硝基化合物
-NO₂
~210(强)
~270(10~20)
亚硝酸酯
-ONO
~220(2000)
~350(0~80)
硝酸酯
-ONO₂
--
~270(10~20)
2-丁烯醛
CH₃=CHCH=O
~217(16,000)
321(20)
联乙酰
O=CH-CH=O
--
435(18)
2,4-己二烯醛
CH₃=CHCH=CHCH=O
~263(27,000)
--
从上表可以看出n→π*跃迁的值很小,一般是由十到几百κ值小的原因,可以从羰基的轨道结构得到解释(见右图)。从图中羰基的轨道图中看到,n轨道的电子与π电子集中在不同的空间区域,因此,尽管n→π*跃迁需要的能量较低,由于在不同的空间,故n轨道的电子跃迁到π轨道的可能性是比较小的,产生跃迁的概率不大。由于κ值是由电子跃迁的概率决定的,所以n→π*跃迁的κ值很小,这种跃迁称为禁忌跃迁,与n→π*跃迁比较κ值要小2~3个数量级。根据n→π*跃迁显示弱的吸收带,同时根据吸收位置,可以预示某些基团的存在,在结构测定中相当有用。
芳香族化合物
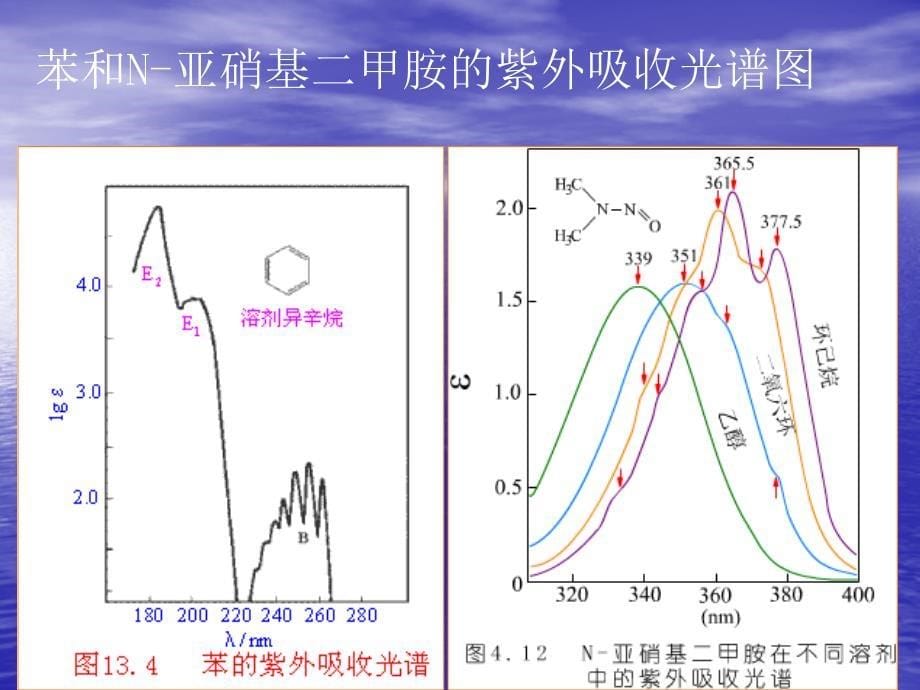
芳香族化合物都具有环状的共轭体系,一般来讲,它们都有三个吸收带。芳香族化合物中最重要的是苯,苯的带Ⅰλmax=184 nm(κ=47000),在真空紫外。带Ⅱλmax=204 nm(κ=6900),带Ⅲλmax=255 nm(κ=230)。下图所示为苯的带Ⅲ在255 nm处的吸收。因为电子跃迁时伴随着振动能级的跃迁,因此将带Ⅲ弱的吸收分裂成一系列的小峰,吸收最高处为一系列尖峰的中心,波长为255 nm,κ值为230,中间间隔为振动吸收,这种特征可用于鉴别芳香化合物。
苯衍生物的带Ⅱ、带Ⅲ亦均在近紫外吸收,下表是苯衍生物的吸收带。
苯衍生物的吸收带 取代基
带Ⅱλmax/nm(κ)
带Ⅲλmax/nm(κ)
H
204(6,900)
255(230)
-NH₃
203(7,500)
254(160)
-CH₃
206(7,000)
261(225)
-I
207(7,000)
257(700)
-Cl
209(7,400)
263(190)
-Br
210(7,900)
261(192)
-OH
210(6,200)
270(1,450)
-OCH₃
217(6,400)
269(1,480)
-CO₂-
224(8,700)
268(560)
-COOH
230(11,600)
273(970)
-NH₂
230(8,600)
287(1,430)
-O-
235(9,400)
287(2600)
-CHO
244(15,000)
280(1,500)
-CH=CH₂
244(12,000)
282(450)
-NO₂
252(10,000)
280(1,000)
注:以上用水、甲醇或乙醇为溶剂。
有些基团的紫外吸收光谱与pH关系很大, 例如苯胺在酸性条件下由于氮上孤电子对与质子结合,它的吸收光谱与苯环类似;如酚在酸性与中性条件下的吸收光谱与碱性时不一样。
影响因素
生色基和助色基
凡是能在某一段光波内产生吸收的基团,就称为这一段波长的生色基(chromophore)。紫外光谱的生色基是:碳碳共轭结构、含有杂原子的共轭结构、能进行n→π*跃迁的基团、能进行n→σ*跃迁并在近紫外区能吸收的原子或基团。常见的生色团列于下表。
常见生色团的吸收峰 生色团
化合物
溶剂
λmax/nm
κmax
H₂C=CH₂
乙烯(或1-己烯)
气态(庚烷)
171(180)
15530(12500)
HC≡CH
乙炔
气态
173
6000
H₂C=O
乙醛
蒸汽
289,182
12.5,10000
(CH₃)₂C=O
丙酮
环己烷
190,279
1000,22
-COOH
乙酸
水
204
40
-COCl
乙酰氯
庚烷
240
34
-COOC₂H₅
乙酸乙酯
水
204
60
-CONH₂
乙酰胺
甲醇
295
160
-NO₂
硝基甲烷
水
270
14
(CH₃)₂C=N-OH
丙酮肟
气态
190,300
5000,-
CH₂=N⁺=N⁻
重氮甲烷
乙醚
417
7
C₆H₆
苯
水
254,203.5
205,7400
CH₃-C₆H₅
甲苯
水
261,206.5
225,7000
H₂C=CH-CH=CH₂
1,3-丁二烯
正己烷
217
21000
孤立的C=C,C≡C的π→π*跃迁的吸收峰都在远紫外区,但当分子中再引入一个与之共轭的不饱和键时,吸收就进人到紫外区,所以该表将C=C,C≡C也算作生色团。
具有非键电子的原子连在双键或共轭体系上,形成非键电子与π电子的共轭,即P-π共轭,使电子活动范围增大,吸收向长波方向位移,并使颜色加深。这种效应,称助色效应,这种基团称为助色基(anxochmme),如一OH,一OR,一NH₂,一NR₂,一SR,卤素等均是助色基。下表为乙烯体系、不饱和羰基体系及苯环体系被助色基取代后波长的增值。
λmax/nm的增值 体系
NR2
OR
SR
Cl
Br
X-C=C
40
30
45
5
-
X-C=C-C=O
95
50
85
20
30
X-C₆H₅ 带Ⅱ
51
20
55
10
10
带Ⅲ
45
17
23
2
6
表中X为助色基。
红与蓝/紫移现象
由于取代基或溶剂的影响,使最大吸收峰向长波方向移动的现象称为红移(red shift)现象。由于取代基或溶剂的影响,使最大吸收峰向短波方向移动的现象称为蓝(紫)移(blue shift)现象。波长与电子跃迁前后所占据轨道的能量差成反比,因此,能引起能量差变化的因素如共轭效应、超共轭效应、空间位阻效应及溶剂效应等都可以产生红移现象或紫移现象。
将烷基引入共轭体系时,烷基中的C一H键的电子可以与共轭体系的π电子重叠,产生超共轭效应,其结果使电子的活动范围增大,吸收向长波方向位移。超共轭效应增长波长的作用不是很大,但对化合物结构的鉴定,还是有用的。下表列举的数据表明了在共轭体系上的烷基对吸收波长的影响。
烷基对共轭体系吸收波长的影响 化合物
λmax/nm
CH₂=CH-CH =CH₂
217
CH₃-CH=CH-CH=CH₂
222
CH₃-CH=CH-CH=CH-CH₃
227
CH₂=C(CH₃)-C(CH₃)=CH₂
227
CH₂=CH=C(CH₃)=O
219
CH₃-CH=CH-C(CH₃)=O
224
(CH₃)₂C=CH-C(CH₃)=O
235
C₆H₆
255
CH₃-C₆H₅
261
由于溶剂与溶质分子间形成氢键、偶极极化等的影响,也可以使溶质吸收波长发生位移。如π→π*跃迁,激发态比基态的极性强,因此极性溶剂对激发态的作用比基态强,可使激发态的能量降低较多,以使基态与激发态之间的能级的能差减小,吸收向长波位移,即发生红移现象。又如n→π*跃迁,在质子溶剂中,溶质氮或氧上的n轨道中的电子可以被质子溶剂质子化,质子化后的杂原子增加了吸电子的作用,吸引n轨道的电子更靠近核而能量降低,故基态分子的n轨道能量降低,n→π*跃迁时吸收的能量较前为大,这使吸收向短波位移,即发生紫移现象,见下图。
由此可见,溶剂对基态、激发态与n态的作用是不同的,对吸收波长的影响亦不同,极性溶剂比非极性溶剂的影响大。因此在记录吸收波长时,需要写明所用的溶剂。紫外中常用的溶剂为水、甲醇,乙醇、己烷或环己烷、醚等。溶剂木身也有一定的吸收带,虽然其κ值小,但浓度一般比待测物的浓度大好几个数量级,因此,如果与溶质的吸收带相同或相近,将会有干扰,选择溶剂时,要予以注意。
增减色效应
使κ值增加的效应称为增色效应(hyperchromic effect)。使κ值减弱的效应称为减色效应(hypochromic cHect)。κ值与电子跃迁前后所占据轨道的能差及它们相互的位置有关,轨道间能差小,处于共平面时,电子的跃迁概率较大,κ值也就较大。在分子中,相邻的生色基由于空间位阻效应而不能很好的 共平面,对化合物的吸收波长及κ值均有影响。例如二苯乙烯由于存在双键,具有顺反异构体,反式异构体的两个苯环可以与烯的键共平面,形成一个大的共轭体系,它的紫外吸收峰在λmax=290 nm(k=27,000);而顺式异构体两个苯环在双键的一边,由于空间位阻不能很好地共平面,共轭作用不如反式的有效,它的紫外吸收λmax=280 nm(κ=14,000)。这种由于空间位阻使共轭体系不能很好共平面而引起的吸收波长与κ值的变化,在紫外吸收光谱中是一种普遍现象,在结构测定中十分有用。
应用范围
医药方面
紫外光谱在破析一系列维生素、抗菌素及天然产物的化学结构曾起过重要作用,如维生素A1、维生素A2、维生素B12、维生素B1、青霉素、链霉素、土霉素、萤火虫尾部的发光物质等。
例如利血平具有两个共轭体系结构,水解得到利血平酸和3,4,5-三甲氧基苯甲酸。利血平酸经LiAlH4还原为利血平醇,其光谱与2,3-二甲基-6-甲氧基吲哚的紫外光谱相似。将合成的利血平醇与3,4,5-三甲氧基苯甲酸的紫外光谱叠加起来所得谱线与利血平的吸收曲线基本吻合,进一步由合成最后确定利血平的结构。[1]
性能测试
光致变色现象是指在光的照射下颜色发生可逆变化的现象,可通过紫外光谱进行测试研究。如螺恶嗪类化合物A的环己烷溶液是没有颜色,但在365nm连续的紫外光的照射下,溶液变成蓝色,在可见区域产生吸收。随照射时间的延长,吸收峰的强度逐渐变大,直至不再变化为止,将化合物的溶液放在暗处,其在可见光区域的吸收会逐渐下降。
光致变色材料作为一类新型功能材料,有着十分广阔的应用前景。例如可以作为光信息存储材料、光开关、光转换器等,这些材料在机械、电子、纺织、国防等领域都大有作为。光致变色涂料、光致变色玻璃、光致变色墨水的研制和开发,具有现实性的应用意义。除了以上的应用,光致变色材料还可以作为自显影感光 胶片、全息摄影材料、防护和装饰材料、印刷版和印刷电路和伪装材料等。
特别要指出的是,光致变色化合物作为可擦重写光存储材料的研究,是近些年来光致变色领域中研究的热点之一。作为可擦写光存储材料的光致变色光存储介质,应满足在半导体激光波长范围具有吸收、非破坏性读出、良好的热稳定性、优良的抗疲劳性和较快的响应速度等条件。