亨廷顿病查看源代码讨论查看历史

{kind=link}

{kind=link}

{kind=link}
亨廷顿病(Huntington disease,HD)是一种以不自主运动、精神异常和进行性痴呆为主要临床特点的显性遗传性神经系统变性病。属于基因动态突变病或多谷酰胺重复病的范畴。因亨廷顿病以舞蹈症状为突出的临床症状,曾将本病命名为大舞蹈病、亨廷顿舞蹈病、慢性进行性舞蹈病或遗传性舞蹈病。
1872年由美国内科医师Huntington对亨廷顿病的临床症状首先进行了描述,1911年Alzheimer对病理改变作了观察,1993年确定其致病基因位于第4对常染色体短臂63位点,此基因编码的蛋白,命名为亨廷素(Huntingtin)。病理改变特点是纹状体和大脑皮质的神经细胞脱失,最近发现在大脑皮质存在泛素阳性神经细胞核内包涵体和营养不良神经突起。
亨廷顿病患者多数发病年龄在25~40 岁,平均发病年龄在40 岁,此病持续5~30 年,平均14 年。5%~10%的患者发病年龄在10~20 岁,1%的患者发病年龄在儿童期,个别患者的发病年龄在80 岁以后。目前没有任何药物可以改变亨廷顿病的自然病程,但可以采取措施改善临床症状、减少舞蹈样动作。治疗集中在对心理与神经征候两方面的症状治疗,同时进行必要的支持治疗。
病因
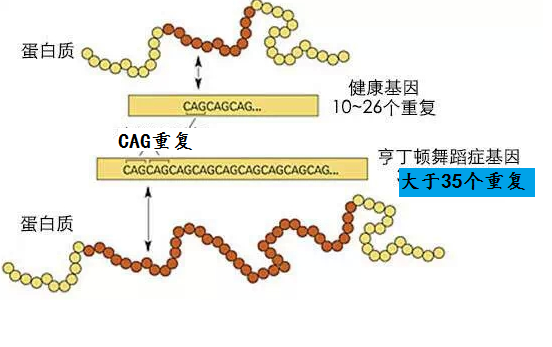
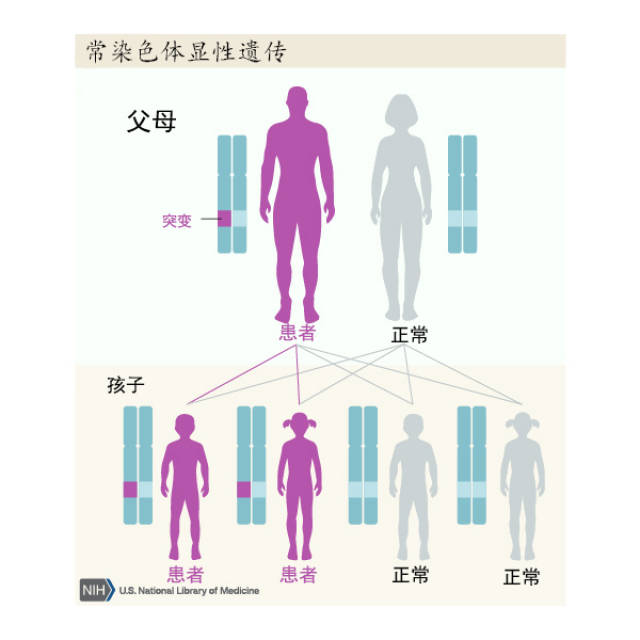
亨廷顿病是影响纹状体和大脑皮质的常染色体显性遗传病,呈完全外显率,受累个体后代50%发病。HD为4号染色体短臂4p16.3的Huntingtin基因突变所致,基因产物为CAG三核苷酸重复扩增产生Huntingtin蛋白,正常人为11~34个CAG重复序列,HD为40个以上。只要了遗传致病基因,或早或晚会出现症状,纯合子与杂合子的临床症状无明显差异,临床亦偶见散发病例。根据发病年龄,HD可分为青年型(20岁前发病)及成年型。
发病机制及病理生理
如果把大脑想成是一颗球,那么在靠近球心的部位 (也就是大脑的深部),有个由众多神经细胞组成的构造叫基底核 (basal ganglia)。它与大脑的运动、情绪、学习、记忆功能有密不可分的关系。就运动功能而言,基底核扮演着大脑秘书的角色,将大脑发出的命令先行处理、修饰过,再把这个命令传回大脑负责运动的部位,然后才把指令传送到身体各处肌肉,指挥我们的一举一动。[1]
亨廷顿病患者体内的异常亨廷顿蛋白首先会影响其脑内的基底核,使得基底核无法修饰或抑制大脑的指令,于是全身肌肉便不受控制地运动,表现为舞蹈样动作。到了疾病的晚期,连负责下达指令的大脑表层也会逐渐死亡,届时病人可能失去所有行动能力,并出现认知功能下降甚至痴呆。*[2]
亨廷顿病患者主要病理改变为基底节区萎缩,其中以尾状核最为明显,壳核和苍白球也有不同程度的萎缩。神经元缺失主要见于基底节区,其中尾状核和壳核的神经元功能障碍与舞蹈样动作有关,皮质神经元缺失可能与痴呆有关。[3]
亨廷顿病的临床表现
亨廷顿病为常染色体显性遗传。子女的发病几率是50%。父系遗传占优势者发病较早,而母系遗传占优势者发病较晚。但如母亲已发病,在妊娠过程中,由于母体与胎儿的相互作用,大部分胎儿流产。而由父系遗传的小孩多能存活。和其他多谷酰胺重复病一样,亨廷顿病的遗传呈现遗传早发现象,即一代比一代发病早,且一代比一代症状重。
亨廷顿病的临床症状包括三方面,即运动障碍、认知障碍和精神障碍,这些临床表现均可以作为首发症状出现。
1、 运动障碍
进行性发展的运动障碍表现为四肢、面、躯干的突然、快速的跳动或抽动,这些运动不可预先知道,也可以表现为不能控制的缓慢运动。查体发现舞蹈样不自主运动和肌张力不全。舞蹈样不自主运动是本病最突出特征,大多开始表现为短暂的不能控制的装鬼脸、点头和手指屈伸运动,类似无痛性的抽搐,但较慢且非刻板式。随病情发展,不随意的运动进行性加重,出现典型的抬眉毛和头屈曲,当注视物体时头部跟着转动,患者行走时出现不稳,腾越步态,加上不断变换手的姿势,全身动作像舞蹈。在疾病后期患者因全身不自主运动而不能站立和行走。即使坐着也不稳,身体扭动,突然站起又突然坐下,卧床后躯干和肢体仍不停的扭动。当病情发展时,随意运动受损愈益明显,动作笨拙、迟缓、僵直,不能维持复杂的随意运动,出现吞咽困难、讲话吞吞吐吐和构音障碍。出现不正常的眼球活动异常。在病的晚期随意运动减慢,呈现出四肢不能活动的木僵状态。多数患者腱反射和感觉正常。
舞蹈样运动障碍是成年型亨廷顿病的典型运动障碍。在20岁前起病的少年型患者(占亨廷顿病的5%~10%)中,以不动性肌强直为主要运动障碍。表现为肌强直、肌阵挛,至晚期则呈角弓反张。此外与成人患者不同,约50%的少年型亨廷顿病者有全身性癫痫发作。
2 认知障碍
进行性痴呆是亨廷顿病患者另一个特征。痴呆在早期具有皮质下痴呆的特征,后期表现为皮质和皮质下混合性痴呆。
认知障碍在亨廷顿病的早期即可出现。开始表现为日常生活和工作中的记忆和计算能力下降,患者记住新信息仅有轻度损害,但信息作修饰以便有效储存有明显困难,回忆也有显著缺陷。由于词的流利性、视空间功能及对社会和人际关系的判断能力下降,病人变的比较混乱,出现人格的改变。
言语的改变,包括口语流利性测验不良,轻度找词困难和构音障碍。口语流利性损害是亨廷顿病最早能计量查出的认知功能不正常之一。在病的中期和晚期,患者不能完成需要组织、连续和语言学精心加工的语言测验,也不能完成需回忆不常用词的命名测试。但这些测试还需要记忆和认识能力,超出了语言范围。没有典型的错语和失语症,但构音和韵律障碍为本病患者的突出特征。舞蹈样运动障碍常可累及舌和唇,破坏了发音的韵律和敏捷性,妨碍了言语的量、速度、节律和短语的长度,使口语呈现一种暴发性质。由于患者仍保留词的识别记忆及对手的识别和对物的命名能力,亨廷顿病患者能继续与人交流。 随病情发展,集中力和判断力进行性受损。患者缺乏启动解决问题的行为。在需要计划和连续安排信息的作业上感到特别困难。视空间能力下降,对结构的判断有困难。在需要连续安排运动的额叶系统测验上,如手的连续变换动作有困难。
3、 精神障碍
首先出现的精神状态变化为人格行为改变,包括焦虑、紧张、兴奋易怒、或闷闷不乐、或不整洁以及兴趣减退,出现反社会行为、精神分裂症、偏执狂和幻觉。情感障碍是最多见的精神症状,且多出现在运动障碍发生之前。由于情感障碍出现在患者的运动障碍出现之前,或了解其家族疾病特点之前,所以不是反应性障碍。此外抑郁症状的发生率也很高,对患者的重度抑郁症状如能早期发现并及时治疗,可预防自杀。亨廷顿病患者的神经和精神性障碍进行性衰退,最后患者处于呆傻、缄默状态。
4、青少年型Huntington舞蹈病
在儿童及青少年期起病,20岁前起病约10%,年龄小于4岁起病约5%。临床表现与成人HD不同,病程进展较快,肌张力障碍是突出表现,常以强直替代舞蹈样运动。尚可见Parkinson综合征、小脑性共济失调、眼球运动异常、肌阵挛及癫痫发作等,可出现精神衰退及行为异常,部分患者表现运动过度。少数病例运动症状不典型(Westphal变异型),表现进行性肌强直和运动减少,舞蹈-手足徐动样症状不明显,多见于儿童期或20岁以前发病者。癫痫和小脑性共济失调也是青少年型常见特点,伴痴呆和家族史可提示诊断。
辅助检查
1、遗传学检测
是确诊重要手段,PCR 法检测亨廷顿基因(TT15)中CAG 重复拷贝数,正常人不超过38 个拷贝,患者在39 个以上,阳性率高,只需检测患者本人,可作到疾病症状前诊断和产前诊断等。
2、脑电图
可有弥漫性异常,无特异性。主要为低波幅快波,尤其额叶明显,异常率占88.9%。α活动减少,波幅降低。视觉诱发电位波幅降低,但首波部分潜伏期正常。患者P100 不正常,检测P300 可作为早期认知功能下降的客观指标。
3、影像学检查
头部CT或MRI对于诊断亨廷顿病具有重要的临床价值,典型的影像学特点是双侧尾状核萎缩,导致侧脑室额角外侧面向外膨起。SPECT 检查发现尾状核和豆状核区域血流明显下降,额叶和顶叶血流也有下降,与患者这些部位的病理改变有关。PET 表现尾状核区葡萄糖代谢明显降低,尾状核区的代谢活性下降可出现在尾状核萎缩前。
临床诊断标准
1.典型HD的家族史。
2.非其他因素导致的进行性运动异常伴舞蹈和僵直。
3.非其他因素导致的精神障碍伴随进行性痴呆。
影像学检查发现对称性尾状核萎缩可以进一步支持亨廷顿病的诊断。在有症状的亨廷顿病患者中,已知左旋多巴可以使舞蹈样动作增加,左旋多巴可引起舞蹈样动作的患者比不引起舞蹈样动作者更可能发生本病,可以诱发处于亚临床状态的患者出现临床表现,用于早期诊断,该试验存在一定的假阴性反应,阴性结果不能完全除外发病的可能性。PET检查发现尾状核部位的葡萄糖代谢减低,也可以出现在亚临床状态的患者,可用作超早期诊断。在亚临床患者如果基因检查发现亨廷素基因(TT15)三核苷酸串联重复序列异常扩展超过40可以进一步确定诊断。由于亨廷顿病具有完全外显的常染色体显性遗传特点,因此亨廷顿病的早期基因诊断具有重要意义,为产前诊断和遗传咨询提供可靠的依据。[4]
鉴别诊断
多数亨廷顿病患者有家族史,但通过基因检查手段也发现一些散发患者,所以需与其他类型的遗传性和散发性舞蹈病进行鉴别。在家族性疾病中齿状核-红核-苍白球-丘脑下核萎缩、良性遗传性舞蹈病和家族性棘红细胞增增多增多症具有类似的临床特点。散发性舞蹈病主要包括药物性、妊娠性、血管疾病、甲状腺功能亢进型、系统性红斑狼疮、狼疮抗凝固综合征、红细胞增多症、艾滋病和风湿性舞蹈病。对患者进行详细的临床检查和必要的辅助检查有助于亨廷顿病的鉴别诊断。
1、 良性家族性舞蹈症
良性家族性舞蹈症是一种常染色体显性、隐性和性连锁的中枢神经系统疾病,分为婴儿早期、儿童期和少年早期三种类型,典型临床症状为非进行性的舞蹈表现,和亨廷顿病不同之处在于智能和精神均正常,影像学检查均无明显异常改变,基因检查发现早期发病者的基因位于常染色体14p可以采用多巴胺受体拮抗药进行治疗,近来此病是否为一个独立的疾病还是一个疾病综合征受到疑问。
2、 风湿性舞蹈病
风湿性舞蹈病是一种散发的良性自限性疾病,病理改变主要表现为基底核炎性病变,主要发病时间在5~15岁,11岁后女性较多。起病多有精神异常,而后隐匿出现不自主的运动,多涉及面部,可伴有构音障碍和吞咽困难,不自主运动更为唐突、暴发,跳动样和抽动样,与亨廷顿病的舞蹈样运动、非刻板模式不同,有些儿童出现肌张力低下,痴呆则罕见。首次发病后持续时间不超过6个月,但25%的患者在发病2年后有复发。部分患者可以伴随出现风湿热、心肌炎和关节炎,血沉快或抗链球菌溶血亲“O”滴度可增高。影像学检查无异常改变。早期可以应用青霉素和激素治疗治疗,但不能缩短舞蹈病的自然病程。
3、神经棘红细胞病
神经棘红细胞病是一种伴随中枢神经系统和周围神经损害的隐性遗传性疾病,其特征为进行性神经退行性变,伴舞蹈样动作及棘形红细胞增增多。根据遗传方式分为常染色体隐性或显性遗传的舞蹈病-棘形红细胞增增多增多症,以及 X-连锁Mcleod综合征两种类型。临床表现与亨廷顿病有许多共同特点。此症多于15~35岁、以肢体和躯干的舞蹈以及口面运动障碍开始发病,也可以出现肌张力不全和帕金森综合征的表现,常合并周围神经病。运动障碍持续进行导致病残,于50~70岁死亡。患者可以出现严重的行为障碍和情绪改变,但痴呆不明显。头颅CT检查显示纹状体萎缩,特别是尾状核头部萎缩最明显。血涂片检查发现外周血的红细胞为棘红细胞。血清肌酸磷酸激酶和乳酸脱氢酶含量可增高。肌电图和肌肉活检有神经原性肌萎缩。神经病理检查和亨廷顿病相似,尾状核和壳核萎缩,小细胞消失、大神经元保存,但没有泛素和亨廷素阳性的神经细胞核内包涵体。临床上,神经棘红细胞增增多增多增多症与亨廷顿病的区别是:隐性遗传、无明显痴呆、有周围神经病和神经元性肌萎缩、棘红细胞增增多、病理改变没有亨廷素阳性的神经细胞核内包涵体。
4、 其他类型的舞蹈病
药物性迟发性运动障碍出现在精神病患者长期应用精神阻滞药后,最显著的动作累及口和舌,但手、腿、躯干和呼吸肌也可发生舞蹈手足徐动症,智能障碍仅出现在部分患者的晚期。此病的诊断主要依靠长期应用精神阻滞药的药物史。妊娠性、血管疾病、甲状腺功能亢进、系统性红斑狼疮、狼疮抗凝固综合征、红细胞增多症可以出现舞蹈症表现,这些疾病均存在相应的内科表现,注意观察相关的内科症状其鉴别诊断不困难。
治疗
目前没有任何药物可以改变亨廷顿病的自然病程,但可以采取措施改善临床症状、减少舞蹈样动作。治疗集中在对心理与神经征候两方面的症状治疗,同时进行必要的支持治疗。要让患者及可能得病者树立信心,相互帮助,建成富有乐观主义的家庭。
亨廷顿病患者脑内γ-氨酪酸(GABA)减少,胆碱能活动受抑制,而多巴胺活动过度,可选用对抗多巴胺能药物或多巴胺受体抑制剂。
1、对抗多巴胺能药物或多巴胺受体抑制剂
丁酰苯类药物中的氟哌啶醇和吩噻嗪类药物氯丙嗪、奋乃静等是主要治疗药物,可以阻滞多巴胺受体。苯酰胺类药物如硫必利(泰必利)有抗多巴胺能的作用,3次/d,100mg/次。
2、 提高胆碱的含量
毒扁豆硷抑制中枢胆碱酯酶的活性,阻止胆碱的降解,可改善舞蹈样运动。
3、 增加中枢神经系统的γ-氨酪酸(GABA)含量
异烟肼是γ-氨基丁酸转移酶的抑制剂,可能使中枢的γ-氨酪酸(GABA)含量升高,使有些患者有轻到中度的进步。一般剂量为10~20mg/kg,每一疗程用药时间4个月~1年;同时应用维生素B6效果更好。
4、 痴呆症状治疗
目前还没有很好的药物。但精神症状通过药物治疗可以获得改善。可用阿米替林、多塞平(多虑平)改善患者的抑郁症状。对暴躁和愤怒暴发时可用氟哌啶醇和碳酸锂联合治疗。
5、 神经细胞移植或胚胎纹状体组织的移植
尚处于探索之中,是否有效还不能确定。
6、其他治疗
可配合应用神经系统促代谢药物、维生素类和能量合剂等。抗自由基治疗、抗氧化和抗细胞兴奋毒性治疗可能也具有一定的疗效。此外加强肢体功能训练和进行心理治疗也可以获得良好的疗效。[5]
预后
亨廷顿病通常持续10~20年,病后15~16年死亡,女性患者病程较长。
视频
亨廷顿病(医学视频)
2分钟神经科学 - 亨廷顿病
亨廷顿舞蹈症可以治疗
参考资料
- ↑ 吴志英. 亨廷顿病的分子发病机制及治疗进展. 中国现代神经疾病杂志. 2009, 9 (3): 238-241.
- ↑ 变异蛋白在血细胞中逐渐积累引发亨廷顿病. 2012-9-22 [07 三月 2020] (中文).
- ↑ 中华医学会神经病学分会帕金森病及运动障碍学组. 亨廷顿病的诊断与治疗指南. 中华神经科杂志. 2011, 44 (9): 638-641.
- ↑ 莫亚勤; 李麓芸; 卢光琇. 亨廷顿病的基因诊断. 遗传. 2005, 27 (6): 861-864.
- ↑ 中华医学会神经病学分会帕金森病及运动障碍学组. 亨廷顿病的诊断与治疗指南. 中华神经科杂志. 2011, 44 (9): 638-641.