中国首个选择性RET抑制剂普拉替尼查看源代码讨论查看历史
 |
中国首个选择性RET抑制剂普拉替尼RET(转染重排基因)融合阳性的非小细胞肺癌(NSCLC)为肿瘤驱动基因异常激活导致的恶性肿瘤[1],以含铂化疗和毒性较大的多靶点激酶抑制剂(MKI)治疗为主,临床上急需通过精准疗法选择性靶向RET变异和预期的耐药突变,来提供持久的临床获益。
普拉替尼是RET激酶及致癌性RET突变体的一种强效、选择性抑制剂。可选择性抑制RET激酶活性,可剂量依赖性抑制RET及其下游分子磷酸化,有效抑制表达RET(野生型和多种突变型)的细胞增殖。普拉替尼对野生型(WT)RET以及致癌性RET突变和融合蛋白的抑制作用具有量效关系,对酶活性的半数最大抑制浓度(IC50)在亚纳摩尔范围内。在细胞系统中,普拉替尼在低纳摩尔效价下即可抑制致癌性RET突变体和RET融合蛋白的激酶活性。并且普拉替尼对RET具有高度选择性,高于对其他450多种激酶的选择性。
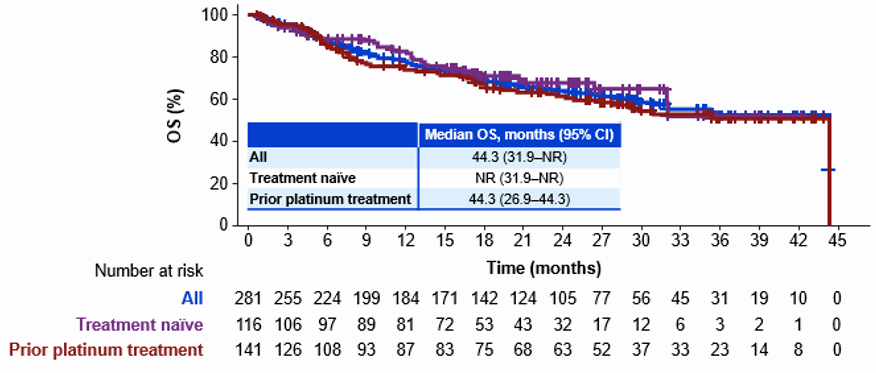
在一项多中心、非随机、开放性、多队列临床研究ARROW中(NCT03037385)评估了本品治疗RET融合阳性的转移性NSCLC患者的有效性。由于明显优于传统治疗药物的疗效和良好的安全性,普拉替尼在II期结束后就获得FDA附条件加速批准上市。并且,基于中国NSCLC桥接队列同样优异并且与全球人群高度一致的疗效数据,普拉替尼胶囊获得NMPA药品审评中心(CDE)优先审评资格,于2021年3月23日获得NMPA附条件批准上市。
普拉替尼是目前中国首个且唯一获批治疗既往接受过含铂化疗的RET融合阳性NSCLC的选择性RET抑制剂,使得RET靶向治疗成为继EGFR、ALK、ROS1等肺癌靶点治疗后的另一个巨大突破。
案例关键词:非小细胞肺癌[2](NSCLC)、普拉替尼、RET、靶向治疗
研发背景、设计、历程及相关新技术应用情况
受体酪氨酸激酶RET(转染重排基因)的异常激活是导致多种实体肿瘤生长和增殖的关键驱动因子。RET基因重排导致的融合蛋白在多种实体肿瘤中都有发现,以非小细胞肺癌(NSCLC)最为常见。在选择性RET抑制剂出现之前,RET融合驱动的NSCLC的标准治疗主要为含铂化疗,与驱动基因阴性患者治疗手段相似,也常见毒性较大的多靶点激酶抑制剂(MKI)治疗。免疫治疗在RET融合阳性患者中的缓解率较低,效果不佳。因此,对于经检测证实携带RET融合的晚期NSCLC患者,目前现有的治疗选择无法提供其他致癌基因(如EGFR、ALK)驱动的NSCLC患者所获得的疗效。临床上急需通过精准疗法选择性靶向RET变异和预期的耐药突变,来提供持久的临床获益。
新颖性
由于明显优于传统治疗药物的疗效和良好的安全性,普拉替尼胶囊获得NMPA药品审评中心(CDE)优先审评资格。2021年3月23日,普拉替尼在II期结束后就获得NMPA附条件批准上市。并且,2020年9月4日普拉替尼首先在美国获FDA批准上市后同一个月内,已于2020年9月29日在海南乐城医疗先行区博鳌超级医院实现首例患者特许用药,治疗RET融合阳性的晚期转移性非小细胞肺癌。普拉替尼具有新机制、新靶点、新结构,是中国上市的首个高选择性、强效RET抑制剂。
研发思路
肺癌是全球癌症死亡的最常见原因。大多数NSCLC患者表现为晚期不可切除疾病(ESMO Guidelines 2019),如果不治疗,预计这些患者将在诊断后平均9.4个月内死亡;所有肺癌患者在诊断后5年以上的存活率仅为18%。肺癌的两个主要类别是NSCLC(约占肺癌的85%)和小细胞肺癌。基因变异作为致癌性驱动因子和肺癌治疗疗效的预测因子发挥着关键作用,在过去的20年中,基因检测的发展实现了对基因变异的识别。约75%的肺腺癌携带促进RTK/RAS/RAF信号通路的基因变异,包括KRAS、EGFR、ALK、ROS1、BRAF、MET、NTRK和RET等驱动因子。
在1-2%的NSCLC患者中观察到携带各种致癌性RET重排。通常,RET重排可生成编码融合蛋白的嵌合转录本,这类融合蛋白由RET激酶结构域与具有二聚化结构域的蛋白(如KIF5B、CCDC6、NCOA4)偶联后,继而生成一种组成性活性激酶,促进肿瘤形成。与携带间变性淋巴瘤激酶(ALK)和ROS1-重排的NSCLC相同,携带RET重排的NSCLC通常也是腺癌(偶尔为鳞癌),见于年轻、不吸烟患者。
已知携带RET重排的NSCLC患者使用MKI(卡博替尼、凡德他尼、索拉非尼和艾乐替尼)的初始病例报告和单组研究均已证实其临床活性。这表明,在NSCLC患者中,RET可能是一个有效的靶点。在这些早期研究中,尽管已观察到良好的缓解率,但是缓解持续时间通常较短。MKI治疗可导致显著的毒性反应,因此,需要停止用药和/或调整用药剂量,这也导致无法使用有效抑制RET需要的暴露量。因此,临床上仍需要新的治疗方法,尤其是在体内可选择性并强效抑制RET的精准治疗药物。
技术特色
生化分析结果显示,普拉替尼对野生型(WT)RET以及致癌性RET突变和融合蛋白的抑制作用具有量效关系,对酶活性的半数最大抑制浓度(IC50)在亚纳摩尔范围(0.33-0.45 nM)内。
在细胞系统中,普拉替尼在低纳摩尔效价下即可抑制致癌性RET突变体和RET融合蛋白的激酶活性。在多个RET驱动的模型,包括RET C634W突变体驱动的MTC异种移植瘤模型、表达CCDC6-RET融合的结直肠癌患者源性异种移植瘤(PDX)模型和KIF5B-RET融合驱动的NSCLC PDX模型中,均证实普拉替尼在体内具有剂量依赖性抗肿瘤疗效。普拉替尼的抗肿瘤疗效与药物的暴露量和对肿瘤生物标志物的药效学调节作用(包括对RET活性的直接抑制作用)相关。在小鼠中,10 mg/kg 每日两次(BID)至30 mg/kg BID和60 mg/kg每日一次(QD)的剂量范围内重复给药后,结果显示,普拉替尼具有良好的耐受性,并且可导致肿瘤生长被完全抑制或肿瘤消退。在这些模型中,以肿瘤生长完全抑制时需要的剂量给药时,观察到RET激酶的活性下降了90%。
在检测普拉替尼对RET和其他人体靶点的选择性时,结果显示,普拉替尼对RET具有高度选择性,高于对其他450多种激酶的选择性。普拉替尼对RET的选择性是对VEGFR2和FGFR1的25倍以上,这两种激酶可被多种MKI抑制。在对55个跨膜或可溶性受体、离子通道和单胺转运蛋白进行检测时,普拉替尼也显示出高度选择性。
参考文献
- ↑ 恶性肿瘤是如何形成的?可能和3点有很大的关系,早看早预防 ,搜狐,2021-09-03
- ↑ 非小细胞肺癌的治疗方法有哪些?,搜狐,2022-11-07