溶酶體貯積症檢視原始碼討論檢視歷史
 |
溶酶體貯積症是中國的一個科技名詞。
漢字是世界上最古老的文字之一[1],已有六千多年的歷史。從倉頡造字的古老傳說到公元前1000多年前甲骨文的發現,漢字有着深厚的歷史底蘊。後來的演變經歷了幾千年的漫長曆程,在形體上逐漸由圖形變為筆畫,象形[2]變為象徵,複雜變為簡單;在造字原則上從表形、表意到形聲。
名詞解釋
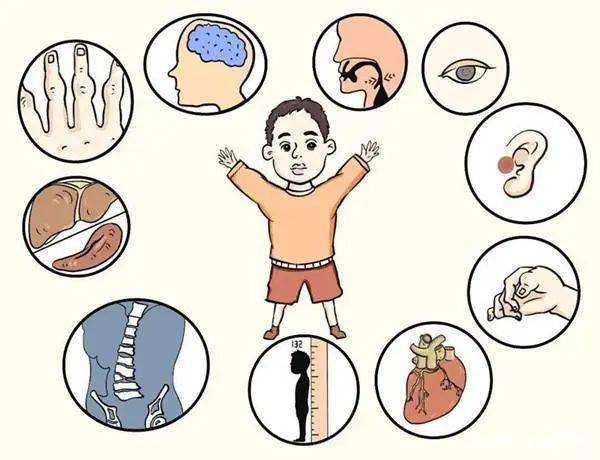
溶酶體貯積症(LSD)是一組遺傳性代謝疾病,是由於基因突變致溶酶體中有關酸性水解酶缺陷,導致機體中相應的生物大分子不能正常降解而在溶酶體中貯積,引起細胞組織器官功能的障礙。
溶酶體貯積症的特點是基於貯積物的複雜性及其組織分布與積聚速度的不同。
溶酶體貯積症既可導致多種系統的病變,也可僅限於神經系統,自出生至成年期均可發病。臨床表現變異大,病情呈進行性加重。絕大多數溶酶體貯積症以常染色體隱性方式遺傳,其中的每一種病發病率雖低,但作為一組病來說就是常見的人類遺傳病之一。
這組疾病多數無有效的治療方法,但可以通過測定溶酶體酶的活性進行病例診斷和產前診斷,防止患兒出生。
我國常見的溶酶體貯積症
黏多糖貯積症
本病是由於酸性黏多糖這種大分子降解過程中所需要的酶缺乏所致,臨床上分為7型,一共有11種酶,除MPS-ⅢC、MPS-ⅢD和MPS-IX外均可檢測,MPS-Ⅱ為X-連鎖隱性遺傳,其餘為常染色體隱性遺傳。
黏脂質貯積症
黏脂質貯積症是由於溶酶體酶磷酸化及定位缺陷而不能轉運入溶酶體導致多種溶酶體酶分泌到細胞外所引起的疾病,是少見的常染色體隱性遺傳疾病,臨床可分為Ⅱ、Ⅲ、Ⅳ三種亞型。實驗室酶學檢測可見多種溶酶體酶活性顯著升高,是正常人酶活性10~30倍。
神經鞘脂貯積症
是指鞘脂降解所需的溶酶體酸性水解酶缺陷或缺少了神經鞘脂激活蛋白,造成了不同的鞘脂如腦苷脂、神經節苷脂或鞘磷脂在溶酶體中貯積,引起中樞神經系統及其他組織病變。法布雷病為X-連鎖隱性遺傳,其餘為常染色體隱性遺傳。
寡糖貯積症
是由於糖蛋白和糖脂中的碳水化合物降解所需的溶酶體酸性水解酶缺乏,造成不同的糖苷脂的貯積所致。
糖原貯積症Ⅱ型
臨床上分為11個亞型,只有糖原貯積症Ⅱ型屬於溶酶體貯積症,是由酸性alpha;-葡糖苷酶缺乏所致。此病採用皮膚活檢,培養皮膚成纖維細胞,檢測皮膚成纖維細胞中的alpha;-葡糖苷酶活性來診斷,是溶酶體貯積症實驗室檢測中唯一不能採用外周血檢測的類型(外周血的該酶檢測方法正在建立中)。
實驗室檢查
溶酶體貯積症的實驗室檢測方法包括:
(1)尿甲苯胺蘭篩查:作為MPS的初篩,若陽性則進行MPS相關的缺陷酶檢測;若陰性可排除MPS,結合臨床應考慮ML的可能。
(2)酶學檢測:這是診斷的金標準,具體酶與疾病的關係見上述。
(3)基因分析:尋找基因突變的情況,可用於檢測雜合子,同時基因分析結合酶學檢測可提高產前診斷的準確性。
產前診斷
由於溶酶體貯積症多數無有效的治療方法,這種患兒的出生無疑給社會和家庭帶來了沉重的經濟和精神負擔,特別是有些患兒發病早,病情進展快,存活期短。這樣的家庭就迫切需要再生育,但再生育又有很高的風險,如何避免風險,對他們進行必要的生育指導,成為溶酶體貯積症患兒家屬的急迫需求。
產前診斷的要求包括兩方面:
(1)先決條件:準確的診斷必須明確到哪一種酶的缺陷。另外,還應注意部分病例的致病原因是與酶活性有關的激活蛋白的缺陷,這種情況下不能用測酶的方法進行產前診斷,只能通過基因分析解決。
(2)取材時間:孕早期11~13周取絨毛(MPSⅠ、ML除外),直接檢測,2周出報告;孕中期16~20周取羊水,經培養後檢測,3周出報告
國內有多家實驗室能夠提供溶酶體酶的檢測,包括北京協和醫院、上海新華醫院、北京中科醫學檢驗所等單位。臨床醫生可以推薦溶酶體貯積症的患者到上述醫院檢測。總之,產前診斷是防止患兒出生的唯一有效措施,也是提高我國的人口素質的一項重要工作。
溶酶體貯積症病例診斷及產前診斷
(一)概述:溶酶體是一種細胞器,即細胞內的超微結構,為單層包被的囊泡,外面是一層脂蛋白膜。它是細胞的處理與回收系統。內部液體呈酸性,含有60多種酸性水解酶,可降解各種生物大分子,如核酸、蛋白質、脂質、粘多糖及糖原等。組成細胞的各種生物大分子都處於動態平衡中,不斷被分解又不斷被再合成。通過內吞作用攝入的生物大分子也需要分解成不同的組分後,才能被利用。這些大分子的分解都是在溶酶體中進行的。
溶酶體中的每一種酶皆有各自的編碼基因。每一種酶的缺陷直接導致某一特定的生物大分子不能正常降解而在溶酶體中貯積。其共同結果都是溶酶體隨之發生腫脹,細胞也變得臃腫失常,細胞功能受到嚴重影響,最終導致疾病,稱為溶酶體貯積症(Lysosomal storage disease, LSD)。酶缺陷的直接原因是編碼基因的突變,絕大多數為常染色體隱性遺傳,是一組較常見的遺傳性代謝疾病。據統計,每1萬個新生兒中將近5例為此病患兒。尚無有效的治療方法。在遺傳性代謝疾病產前診斷中此病所占的比例最大。因此,對高危孕婦進行產前診斷是防止患兒出生的唯一有效措施。
(二)溶酶體酶的共同特性:
1. 皆為酸性水解酶,是含甘露糖的糖蛋白。酶作用最適pH為3.5~5.5。溶酶體依靠質子泵的作用維持酸性環境(pH 5.0~5.5)。
2. 是一種端切酶。溶酶體內的大分子降解時,酶從大分子的末端起,一個個切除糖基、硫酸基、脂及酸殘基等。
3. 以多種形式存在於溶酶體中。多數溶酶體酶存在於可溶性基質中,實驗步驟中不需使用去污劑;少數是溶酶體膜的組成部分(如葡萄糖腦苷脂酶、酸性磷酸酶),因此測酶活性時需用去污劑。
(三)溶酶體貯積症的特點:
1. 絕大部分為常染色體隱性遺傳病。即父母雙方皆為表型正常的雜合子,所生子女出現疾病的風險為四分之一。已發現有30多種疾病。
2. 多種組織或器官受累。主要臨床表現為智力低下、骨骼及神經系統發育障礙。嚴重影響患者的生命力。
3. 臨床病情呈進行性加重。如粘多糖貯積症、GM1及GM2神經節苷脂貯積症、異染性腦白質營養不良等疾病,表現為神經系統損害進行性加重。
4. 貯積物為單層溶酶體膜所包裹。
5. 貯積物成分多樣化。
6. 酶活性測定是最後確診本病的可靠依據。
7. 有的病可用酶補充治療。如戈謝病(Gaucher disease);多數疾病無有效的治療方法,但可通過酶活性測定做出診斷及產前診斷。
由於溶酶體貯積症病情嚴重,多數疾病無有效治療方法,預後不良。患兒的出生給社會和家庭帶來沉重的經濟和精神負擔。這種疾病雖無有效治療方法,但多數可在產前清楚判明胎兒是否患病,有的還可在孕早期作出產前診斷,在優生上具有「預防」的意義。因可在臨床上根據明確的產前診斷結果阻止胎兒出生,它不僅是唯一可行的優生措施,而且能減輕家庭及社會的負擔,提高人口素質。
參考文獻
- ↑ 雲端超市•第407期┃「說文解字,中國最古老的一種文字」——篆書研究 主講人:倪文東,搜狐,2022-10-28
- ↑ 為什麼中國人會發明象形文字?,搜狐,2020-10-06