芳香烴檢視原始碼討論檢視歷史
| 芳香烴 | |
|---|---|

芳香烴( Aromatic hydrocarbons ),通常指分子中含有苯環結構的碳氫化合物。是閉鏈類的一種。具有苯環基本結構,歷史上早期發現的這類化合物多有芳香味道,所以稱這些烴類物質為芳香烴,後來發現的不具有芳香味道的烴類也都統一沿用這種叫法。例如苯、二甲苯、萘等。苯的同系物的通式是CnH2n-6 (n≥6)。芳香烴的π 電子數為4n+2 (n為非負整數)。
主要結構
苯的結構和表達
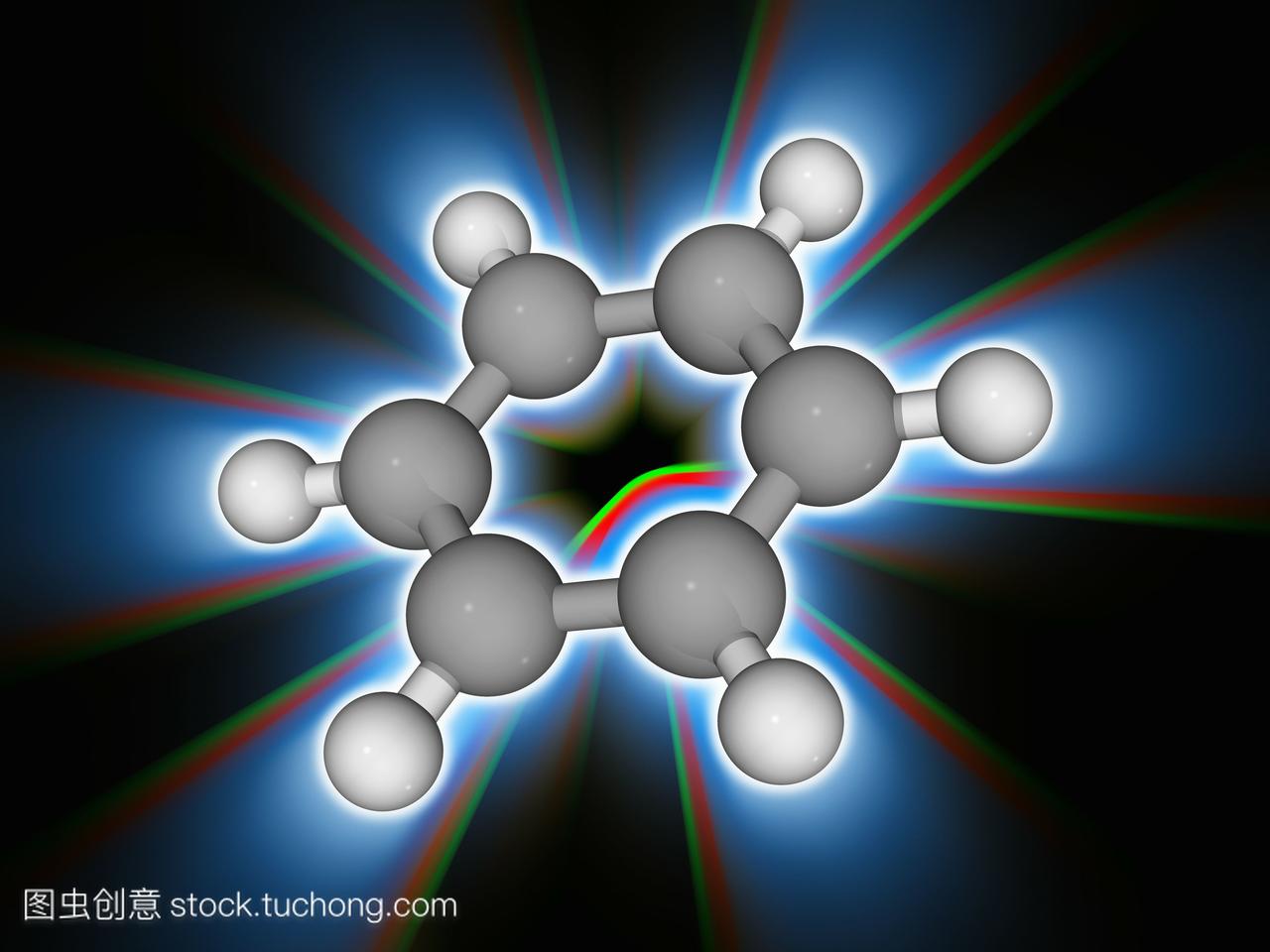
(1)苯的結構
近代物理方法證明:苯分子的六個碳原子和六個氫原子都在一個平面內,因此它是一個平面分子,六個碳原子組成一個正六邊形,碳鍵鍵長是均等的,約為140 pm,介於單鍵和雙鍵之間。碳氫鍵鍵長為108pm,所有的鍵角都為120°。

(2)苯的芳香性
從結構上看,苯具有平面的環狀結構,鍵長完全平均化,碳氫比為1。從性質上看,苯具有特殊的穩定性:環己烯的氫化熱ΔH=-120kJ/mol,1,3-環己二烯的氫化熱ΔH=-232kJ/mol(由於其共軛雙鍵增加了其穩定性)。而苯的氫化熱ΔH=-208kJ/mol。1,3-環己二烯失去兩個氫變成苯時,不但不吸熱,反而放出少量的熱量。這說明:苯比相應的環己三烯類要穩定得多,從1,3-環己二烯變成苯時,分子結構已發生了根本的變化,並導致了一個穩定體系的形成。[1] 苯難於氧化和加成,而易於發生親電取代反應,與普通烯烴的性質有明顯的區別。
苯還具有特殊的光譜特徵。苯環上的氫處於核磁共振的低場。
上述特點說明了苯具有典型的芳香特徵。
(3)苯的表達[2]
怎樣來表達苯的結構?自1825年英國物理學家和化學家Farady M(法拉第)首先從照明氣中分離出苯後,人們一直在探索苯結構的表達式。科學家們提出了各種有關苯結構式的假設;其中比較有代表性的苯的結構式有:

關於苯的結構及它的表達方式已經討論了140多年了,雖然提出了各種看法,但還沒有得到滿意的結果,需要作進一步討論。
聯苯的結構
最簡單的聯苯是二聯苯。在二聯苯中,每個苯環都保持了苯的結構特性。連接兩個苯環之間的單鍵可以自由旋轉,但當二聯苯的四個鄰位氫原子都被相當大的基團取代時,單鍵的旋轉將會受到阻礙,並產生出一對光活性異構體。
物理性質
芳香烴不溶於水,但溶於有機溶劑,如乙醚、四氯化碳、石油醚等非極性溶劑。一般芳香烴均比水輕;沸點隨相對分子質量升高而升高;熔點除與相對分子質量有關外,還與其結構有關,通常對位異構體由於分子對稱,熔點較高。一些常見芳香烴的物理性質列於下表中:

一些常見的芳香烴的名稱及物理性質
化學性質
加成反應
1.苯的加成反應
苯具有特殊的穩定性,一般不易發生加成反應。但在特殊情況下,芳烴也能發生加成反應,而且總是三個雙鍵同時發生反應,形成一個環己烷體系。如苯和氯在陽光下反應,生成六氯代環己烷。
只在個別情況下,一個雙鍵或兩個雙鍵可以單獨發生反應。
2.萘、蒽和菲的加成反應
萘比苯容易發生加成反應,例如:在不受光的作用下,萘和一分子氯氣加成得1,4二氯化萘,後者可繼續加氯氣得1,2,3,4-四氯化萘,反應在這一步即停止,因為四氯化後的分子剩下一個完整的苯環,須在催化劑作用下才能進一步和氯氣反應。1,4-二氯化萘和1,2,3,4-四氯化萘加熱可以失去氯化氫而分別得1-氯代萘和1,4-二氯代萘。
由於稠環化合物的環十分活潑,因此一般不發生側鏈的鹵化。
蒽和菲的9、10位化學活性較高,與鹵素的加成反應優先在9、10位發生。

還原反應
1.Birch還原反應
鹼金屬(鈉、鉀或鋰)在液氨與醇(乙醇、異丙醇或二級丁醇)的混合液中,與芳香化合物反應,苯環可被還原成1,4-環己二烯類化合物,這種反應叫做Birch(伯奇)還原。例如,苯可被還原成1,4-環己二烯。
Birch還原反應與苯環的催化氫化不同,它可使芳環部分還原生成環己二烯類化合物,因此Birch還原有它的獨到之處,在合成上十分有用。
萘同樣可以進行Birch還原。萘發生Birch還原時,可以得到1,4二氫化萘和1,4,5,8-四氫化萘。

2.催化氫化反應
苯在催化氫化( catalytic hydrogenation)反應中一步生成環己烷體系。萘在發生催化加氫反應時,使用不同的催化劑和不同的反應條件,可分別得到不同的加氫產物。蒽和菲的9、10位化學活性較高,與氫氣加成反應優先在9、10位發生。
3.用金屬還原
用醇和鈉也可以還原萘,溫度稍低時得1,4-二氫化萘,溫度高時得1,2,3,4-四氫化萘。
氧化反應
1.苯及其衍生物的氧化
烯、炔在室溫下可迅速地被高錳酸鉀氧化( oxidation),但苯即使在高溫下與高錳酸鉀、鉻酸等強氧化劑同煮,也不會被氧化。只有在五氧化二釩的催化作用下,苯才能在高溫被氧化成順丁烯二酸酐。
烷基取代的苯易被氧化,但一般情況下,氧化時苯環仍保持不變,只是和苯環相連的烷基被氧化成羧基。

而且,不管側鏈多長,只要和苯環相連的碳上有氫,氧化的最終結果都是側鏈變成只有一個碳的羧基,如果苯環上有兩個不等長的側鏈,通常是長的側鏈先被氧化。
只有苯環和一個三級碳原子相連或與一個極穩定的側鏈相連時,在強烈的氧化條件下,側鏈才得以保持,苯環被氧化成羧基。
2.萘、蒽和菲的氧化反應
萘比苯易氧化,在室溫用三氧化鉻的醋酸溶液處理得1,4-萘醌。若在高溫和五氧化二釩的催化下被空氣氧化,則得重要的有機化工原料鄰苯二甲酸酐。
當萘環上有取代基時,活化基團常常使氧化反應在同環發生,而鈍化基團使氧化反應在異環發生。
由於萘環比側鏈更易氧化,所以不能應用側鏈氧化法來製備萘甲酸。
蒽和菲的氧化反應首先在9、10位發生。蒽用硝酸或三氧化鉻的醋酸溶液或重鉻酸鉀的硫酸溶液氧化生成9,10 -蒽醌,9,10-蒽醌是合成蒽醌染料的重要中間體。菲用上述氧化劑氧化生成9,10 -菲醌。

取代反應
芳香族化合物芳核上的取代反應從機理上講包括親電、親核以及自由基取代三種類型。所謂芳香親電取代(aromatic electrophilic substitution)是指親電試劑取代芳核上的氫。苯的親電取代稱為苯的一元素電取代,一元取代苯再在苯環上發生親電取代稱為苯的二元親電取代。典型的芳香親電取代有苯環的硝化、鹵化、磺化、烷基化和酰基化。這些反應的反應機理大體是相似的。
硝化反應
有機化合物分子中的氫被硝基(-NO2)取代的反應稱為硝化反應。苯在濃硝酸和濃硫酸的混合酸作用下,能發生硝化反應,反應的結果是苯環上的氫被硝基取代。
芳香族化合物的硝化反應是一個十分有用的取代反應。例如:苯甲醛的硝化產物間硝基苯甲醛是生產強心急救藥阿拉明的重要原料。
因為醛基易氧化,因此反應必須在低溫(0℃)進行,操作時,先在濃硫酸中加入少量發煙硝酸,冷卻至0℃,然後慢慢滴加苯甲醛和發煙硝酸,反應完成後,立即將產物傾倒在冰中。許多硝基化合物是炸藥。廣泛使用的強烈炸藥TNT是2,4,6-三硝基甲苯,它是甲苯經分階段硝化製備的,即三個硝基是在多次硝化反應中逐步引入的。
三次硝化的硝化試劑(即混合酸)濃度逐漸增高,在生產中,為節約成本,可把第三階段硝化後的混合酸用於第二階段硝化,第二階段硝化後的混合酸用於第一階段硝化。如果需要得到中間產物,反應可以在第一階段或第二階段中止,鄰硝基甲苯和對硝基甲苯可以通過減壓蒸餾或重結品分離提純而分別獲得,2,4二硝基甲苯也能通過重結晶提純得到。
定位效應
一元取代苯進行二元硝化時,已有的基團對後進入基團進入苯環的位置產生制約作用,這種制約作用即為取代基的定位效應(directing effect)。取代基的定位效應是與取代基的誘導效應、共軛效應、超共軛效應等電子效應有關的。
1.取代基的誘導效應和共軛效應
誘導效應與原子的電負性有關。比碳電負性強的原子或基團能使苯環上的電子通過σ鍵向取代基移動,即具有吸電子的誘導效應。電負性比碳弱的原子或基團使取代基上的電子通過σ鍵向苯環移動,即具有給電子的誘導效應。

共軛效應是取代基的σ(或π)軌道上的電子云與苯環碳原子的p軌道上的電子云互相重疊,從而使σ(或π)電子發生較大範圍的離域引起的,離域的結果如使取代基的σ電子向苯環遷移則發生了給電子的共軛效應,如使苯環上的π電子向取代基遷移則發生了吸電子的共軛效應。產生給電子共軛效應的取代基有:
-NR2>-OR>-F,-O->-OR,-F>-Cl>-Br>-I
絕大多數取代基既可與苯環發生誘導效應,也可發生共軛效應,最終的表現是兩者綜合的結果。大部分取代基的誘導效應與共軛效應方向是一致的,但有的原子或基團的誘導效應與共軛效應方向不一致。例如,鹵素的電負性比較大,它具有吸電子誘導效應,鹵苯的鹵原子的p軌道與苯環碳上的p軌道平行重疊,鹵原子的孤電子對離域到苯環上,發生給電子的共軛效應,但總的結果是吸電子的誘導效應大於給電子的共軛效應,因此鹵素是吸電子基,它使苯環的電子云密度降低。取代基的綜合電子效應可以從取代苯的偶極矩大小和方向上表現出來。
在烷基苯中,烷基與苯環不發生共軛作用,但烷基的C-H中σ電子與苯的π電子能發生σ-π超共軛作用,烷基的超共軛作用有微弱的給電子能力。
2.硝基苯的硝化反應
硝基苯硝化的反應式及實驗數據如下所示:
硝基苯+發煙硝酸+濃硫酸-95℃->間二硝基苯(93%)+鄰二硝基苯(6%)+對二硝基苯(1%)
將上面的式子與苯的硝化對比,可以得出下述結論:
(1)硝基苯比苯難硝化得多,需要用比較強的條件,例如提高反應溫度、增加酸的濃度等來實現。
(2)硝基苯硝化時,主要得到間位產物,鄰、對位產物極少。
硝基苯比苯難硝化的原因是:苯環的硝化是一個親電取代反應,硝化反應的機理表明:整個反應的關鍵一步是硝基正離子進攻苯環形成中間體碳正離子。在硝基苯中,因氧、氮的電負性均大於碳,因此硝基有吸電子的誘導效應,叉因為硝基的π軌道與苯環的離域π軌道形成一個π-π共軛體系,使苯環的π電子云也向硝基遷移,所以硝基是一個具有強吸電子誘導效應和吸電子共軛效應的取代基。它使苯環的電子云密度有較大程度的下降,這一方面增加了硝基正離子進攻苯環的難度,同時也降低了反應過程中產生的中間體碳正離子的穩定性,所以硝基苯比苯難硝化。

3.甲苯的硝化反應
甲苯硝化的反應式及實驗數據如下所示:
甲苯+濃硝酸+濃硫酸-30℃->鄰硝基甲苯(58%)+對硝基甲苯(38%)+間硝基甲苯(4%)
甲苯完全硝化,可直接得到三硝基甲苯(TNT)。
實驗結果表明:①甲苯比苯容易硝化;②甲苯硝化時,主要得到鄰位和對位產物。
甲苯比苯容易硝化的原因是:甲基具有微弱的給電子超共軛效應,這種超共軛效應使苯環上的電子云密度有所增加,這一方面使硝基正離子更容易進攻苯環,同時也使反應過程中產生的中間體碳正離子的電荷得到分散而穩定。所以甲苯比苯更易硝化。但甲基的給電子能力是很弱的,因此它對苯環的活潑性影響較弱。
4.氯苯的硝化
氯苯硝化的反應式及實驗數據如下所示:
氯苯+濃硫酸+濃硝酸-60~70℃->鄰氯硝基苯(30%)+對氯硝基苯(70%)+間氯硝基苯(極微量)
實驗結果表明:①氯苯比苯難以硝化;②氯苯硝化時主要得到鄰、對位取代產物。
氯苯比苯難以硝化的原因是:氯原子的吸電子誘導效應比給電子共軛效應大,總的結果使苯環上的電子云密度降低,這一方面使硝基正離子不易進攻苯環,另一方面使反應過程中產生的中間體碳正離子更不穩定,反應時過渡態勢能增大,所以氯苯比苯難硝化。
鹵化反應
有機化合物分子中的氫被鹵素(-X)取代的反應稱為鹵化反應。苯在Lewis酸如三氯化鐵、三氯化鋁等的催化作用下能與氯或溴發生苯環上的鹵化反應生成氯苯或溴苯。
鐵粉與氯氣或溴反應可生成三氯化鐵或三溴化鐵,因此也可以用鐵粉代替三氯化鐵、三溴化鐵做催化劑。反應時,首先是鹵素與苯形成π絡合物,光譜和X射線衍射法都已證明了π絡合物的存在。在形成π絡合物時,氯分子的鍵沒有異裂,然後在缺電子的Lewis酸的作用下,氯分子鍵極化,進而發生鍵的異裂,生成活性中間體碳正離子,然後失去氫生成氯苯。
苯的溴化也可直接進行,但速率很慢。
鹵素由於活潑性不同,發生鹵化反應時,反應性也不同。最大的差別是氟太活潑,不宜與苯直接反應,因直接反應時,只生成非芳香性的氟化物與焦油的混合物。大量的苯在四氯化碳溶液中,與含有催化量氟化氫的二氟化氙反應,可製得產率為68%的氟苯。
碘很不活潑,只有在HNO3等氧化劑的作用下才能與苯發生碘化反應,氧化劑可以將反應產生的HI氧化成碘而有利於反應進行。

{kind=link}
【苯酚的鑑別】
羥基是一個強的活化基團,這從下面的實驗事實可以看出:在盛有少量苯酚( phenol)溶液的試管里滴加過量的濃溴水,很快就有三溴苯酚的白色沉澱產生。這個反應可用來鑑別苯酚。
因此製備一溴苯酚通常要在惰性溶劑中進行,惰性溶劑在這裡起稀釋作用,使反應易於控制在一元階段。例如對溴苯酚通常是在二硫化碳溶劑中進行的。
製備對溴苯胺一般都先將苯胺乙酰化,這一方面可以降低氨基對苯環的活化能力,同時因乙酰氨基的空間位阻較大,可以阻止後進入基團進入氨基的鄰位,而得到對位產物,反應完成後,乙酰基可以水解除去。
在光或能產生自由基的物質的作用下,甲苯的鹵化不發生在芳環上而是在側鏈上,甲苯的三個氫可以被逐個取代,反應機理與丙烯中的σ氫鹵化一樣,是自由基型的取代反應。
如果是較長的側鏈,鹵化反應也可以在別的位置發生,但是σ位的選擇性最高,這是因為苯甲型自由基最穩定的緣故。
磺化反應
有機化合物分子中的氫被磺(酸)基(-SO3H)取代的反應稱為磺化(sulfonation)反應,苯及其衍生物幾乎都可以進行磺化反應,生成苯磺酸或取代苯磺酸。
傅-克反應
Friedel(傅瑞德爾)- Crafts(克拉夫茲)反應,簡稱傅一克反應。有機化合物分子中的氫被烷基(-R)取代的反應稱為烷基化反應,被酰基取代的反應稱為酰基化反應。苯環上的烷基化反應和酰基化反應統稱為傅克反應。
1.傅-克烷基化反應
傅一克烷基化反應(Friedel-Crafts alkylation)的反應機理與磺化、硝化類似,首先在催化劑的作用下產生烷基碳正離子,它作為親電試劑向苯環進攻,形成碳正離子,然後失去一個質子生成烷基苯。
鹵代烷、烯烴、醇、環氧乙烷等在適當催化劑的作用下都能產生烷基碳正離子,鹵代烷、烯烴、醇是常用的烷基化試劑。最初用的催化劑是三氯化鋁,後經證明,許多Lewis酸同樣可以起催化作用。
2.傅一克酰基化反應
傅一克酰基化反應(Friedel-Crafts acylation)的反應機理和烷基化是類似的,也是在催化劑的作用下,首先生成酰基正離子,然後和芳環發生親電取代。
常用的催化劑是三氯化鋁。由於AlCl3能與羰基絡合,因此酰化反應的催化劑用量比烷基化反應多,含一個羰基的酰鹵為酰化試劑時,催化劑用量要多於1 mol反應時,酰鹵先與催化劑生成絡合物,少許過量的催化劑再發生催化作用使反應進行。如用含兩個羰基的酸酐為酰化試劑,因同樣原因,催化劑用量要多於2 mol。
氯甲基化反應與Gattermann-Koch反應
1.氯甲基化反應
氯化苄(henzyl chloride)也稱為苄氯,可通過苯與甲醛、氯化氫在無水氯化鋅作用下反應製得,此反應稱為氯甲基化( chloromethylation)反應。苄氯上的氯十分活潑,可以轉化為各種有用的化合物。
2.Gattermann-Koch反應
在Lewis酸及加壓情況下,芳香化合物與等物質的量的一氧化碳和氯化氫的混合氣體發生作用可以生成相應的芳香醛。在實驗室中則用加入氯化亞銅來代替工業生產的加壓方法。因氯化亞銅可與一氧化碳絡合,使之活性增高而易於發生反應。
電取代經驗規律
苯的多元親電取代是指二元取代苯或含有更多取代基的苯衍生物進行親電取代反應,其中最簡單的是二元取代苯的進一步取代。和苯的二元取代一樣,苯環上已有的取代基對新進入苯環的取代基也有定位作用。二元或多元取代苯的定位問題比一元取代苯複雜。總的來說,最終反映出來的定位作用實際上是苯環上已有取代基的綜合作用,若已有取代基的定位作用一致,則它們的作用可以互相加強。
兩個取代基中間的位置一般不易進入新基團。
當已有取代基的定位作用不一致時,可參照下列經驗規則:
(1)多數情況下,活化基團的作用超過鈍化基團的作用。
(2)強活化基團的影響比弱活化基團的影響大。
(3)兩個基團的定位能力沒有太大差別時,主要得到混合物。
巧妙地利用取代基的定位效應,合理地確定取代基進入苯環的先後次序可以有效地合成芳香族化合物。例如,由苯合成鄰硝基氯苯要先氯化後硝化,而合成間硝基氧苯則要先硝化而後氯化。又如,用甲苯製備3-硝基-5-溴苯甲酸時,因為三個取代基互為間位,因此要優先引入間位定位基,即要先氧化,再硝化,最後溴化。而用甲苯製備2,4一二硝基苯甲酸,則要先硝化再氧化。
除取代基的定位效應外,反應溫度、溶劑、催化劑、新進入取代基的極性、體積等眾多因素對取代基進入苯環的位置也都有影響。例如,甲苯在不同溫度下進行磺化,所得產物中各異構體的產率如下所示:
再如溴苯氯化,產物中鄰、對、間位異構體分別為:42%,51%.7%;隨着進入基團體積的增大,鄰位異構體產量減少,對位異構體增多,這主要是空間效應的結果。因此在進行反應和合成時,要全面考慮問題。
親電取代反應
在正常情況下,萘比苯更易發生典型的芳香親電取代反應,硝化和鹵化反應主要發生在α位上。
由於萘十分活潑,溴化反應不用催化劑就可進行,氯化反應也只需在弱催化劑作用下就能發生。
為什麼取代反應主要發生在α位上?共振理論認為:取代基進攻α位形成的碳正離子中間體有兩個穩定的含有完整苯環結構的極限式,而進攻盧位形成的碳正離子中間體只有一個穩定的含有完整苯環結構的極限式,所以前者比後者穩定。顯然,穩定碳正離子相對應的過渡態勢能也相對較低,所以進攻α位,反應活化能較小,反應速率快。
在發生可逆的磺化反應時,進入的位置和外界的條件很有關係。低溫時,口氫先被取代,當溫度升高後,再轉移到較穩定的p位上,這結果表明α-萘磺酸的生成是受動力學控制的,而β-萘磺酸的生成是受熱力學控制的。
上述現象表明,與萘的硝化、鹵化反應一樣,生成α-萘磺酸比生成β-萘磺酸活化能低,低溫條件下提供能量較少,所以主要生成α-萘磺酸。但磺化反應是可逆的,由於,α-磺基與異環的α-H處於平行位置,空阻較大,不穩定,隨着反應溫度升高,α-萘磺酸的增多,α-磺化反應的逆向速率將逐漸增加;另外,溫度升高也有利於提供β-磺化反應所需的活化能,使其反應速率也加大,β-磺基與鄰近的氫距離較大,穩定性好,其逆向反應速率很慢,所以α-萘磺酸逐漸轉變成β-萘磺酸。
萘的酰化反應既可以在α位發生,也可以在β位發生,反應產物與溫度和溶劑很有關係。
一取代萘進行親電反應時,第一取代基(G)也有定位效應,鹵素以外的鄰對位取代基使環活化,因此取代反應主要在同環發生。
如果第一取代基(G)在β位時,有時6位也能發生取代反應,因為6位也可以被認為是G的對位。
間位取代基使環鈍化,因此取代反應主要發生在異環的α位。
但是,磺化和傅一克反應常在6,7位發生,生成熱力學穩定產物。
蒽比苯、萘更易發生親電取代反應,除磺化反應在1位發生外,硝化、鹵化、酰化時均得9-取代蒽,取代產物中常伴隨有加成產物。
菲的9,10的化學活性很高,取代首先在9,10位發生。
此外菲的1,2,3,4,10和5,6,7,8,9是對應的,所以應有五種一元取代產物。
製備方法
來源
煤和石油是製備一些簡單芳香烴如苯、甲苯等的原料。而這些簡單芳香烴又是製備其它高級芳香族化合物的基本原料。當煤在無氧條件下加熱至1000℃,煤分子通過熱分餾產生煤焦油。而從煤焦油中可以產生苯、甲苯、二甲苯、萘和其它芳香化合物。與煤不同,石油中含有大量的烷烴和少量芳香化合物。石油餾分中主要含有環烷烴和鏈烴,將它們轉化為芳香烴的主要方法是重整和芳構化。芳構化是指含六元環的脂環族化合物在鉑、鈀、鎳等催化劑存在下,加熱脫氫,生成芳香族化合物的過程。重整包括鏈烴裂解、異構化、關環、擴環、氫轉移、烯烴吸氫等過程。重整一般都是在催化劑作用下進行的,常用的催化劑有鉑、銅等。石油工業化學中一個重要的反應稱臨氫重整(或稱鉑重整),就是在氫存在下,以鉑為催化劑使分子結構重新安排,例如在Pt,SiO2/Al2O3,500℃,1~4 MPa及H2存在的反應條件下進行反應。臨氫重整反應過程很複雜,因為反應體系是混合物。
傅一克反應作用
傅一克酰基化反應和脫氫反應在合成稠環體系時起着很大的作用。
合成過程中要使用兩次傅一克酰基化反應,第一次生成γ-氧代γ苯基丁酸,經Cemmensen還原後,再進行第二次酰化關環,然後再經一次Clemmensen還原及硒脫氫後得萘。
蒽醌是合成一大類蒽醌染料的重要中間體,它的工業合成方法是經過兩次傅一克酰基化反應得到蒽醌。蒽醌再經還原脫氫後,得到蒽。
用萘和丁二酸酐發生傅一克酰基化反應,萘的1位及2位上都可被酰化,得到兩個異構體,即β-(1-萘甲酰基)丙酸和β-(2-萘甲酰基)丙酸,然後按標準的方法還原、關環、還原、脫氫就得到菲。
γ-芳基丁酸在多磷酸(或85% H2S04)作用下,加熱環化成六元環酮,環酮用鋅汞齊、鹽酸還原後,再用硒加熱脫氫得到多環芳香族化合物。這個合成多環化合物的重要方法稱為Haworth(哈沃斯)反應。用苯合成萘、由萘合成菲的反應實例說明,Haworth反應在合成稠環化合物中十分有用。
蔻合成方法
蔻(coronene)在自然界中不存在,可用多種方法合成。其中一個方法是利用傅一克反應及硒脫氫反應而實現的。用2,7二甲萘通過N一溴代丁二酰亞胺進行苯甲型的溴化,兩個甲基的氫各被一個溴取代,然後用Wurtz反應將兩分子縮合,即得到一個十四元的環狀化合物。在三氯化鋁的作用下即行關環。這步反應的過程和芳香烴被烯烴烷基化類似。最後用硒脫氫即得到蔻。
六苯並蔻(hcxabenzocoronene)是有機材料中應用十分廣泛的一個中間體,利用二苯乙炔與2,3,4,5-四苯基環戊二烯酮的Diels - Alder反應,再脫酮生成六苯基苯,此化合物經FeCl3氧化即町生成六苯並蔻。
主要危害
苯是一種應用廣泛的有機溶劑,是黏合劑、油性塗料、油墨等的溶劑。短時間內吸入大量苯蒸氣可引起急性中毒。急性苯中毒主要表現為中樞神經系統麻醉,甚至導致呼吸心跳停止。長期反覆接觸低濃度的苯可引起慢性中毒,主要是對神經系統、造血系統的損害,表現為頭痛、頭暈、失眠,白血球持續減少、血小板減少而出現出血傾向,甚至誘發白血病。我國規定操作車間內空氣中苯的濃度不得超過40mg·m,居室內空氣中苯含量平均每小時不得超過0.09mg·m.製鞋、皮革加工業、箱包、家具製造中使用的黏膠劑,噴漆、油漆工作中使用的溶劑都含有苯或苯的同系物,因此從事上述職業的人群要加強防範,避免苯中毒。雖然環芳烴對人體有害,但是人類卻對一種原始的烹飪方式情有獨鍾,那就是燒烤(這可能是由於祖先遺傳給我們的行為因子造成的,在有些時候竟然使我們對環芳烴產生一種愉悅感)。
人若長期接觸或吸入稠環芳烴如萘(俗稱衛生球,過去用來驅蚊防霉)等則會致癌。許多稠環芳烴是強烈的致癌物質,如苯並芘等。秸稈、樹葉等物質不完全燃燒形成的煙霧中含有較多的稠環芳烴,我國有些省市已經禁止焚燒樹葉和秸稈。香煙的煙霧中也存在多種稠環芳烴,提醒青少年應珍視生命,遠離香煙煙霧的危害。
發展狀況
國內情況
榮盛石化增資9.6億投建芳烴項目
榮盛石化擬以自有資金9.6億元,增資全資子公司寧波中金石化有限公司,用於投資建設芳烴項目。
增資後,寧波中金石化註冊資本由3980萬元增至10億元。榮盛石化表示,此次增資有利於確保投資項目順利實施,也有利於公司向產業鏈上游拓展,發揮產業鏈優勢。
此外,榮盛石化同意控股子公司逸盛大化,將坐落於大連經濟技術開發區董家溝居住區的城鎮住宅用地使用權,轉讓給關聯公司大連海濱置業有限公司,轉讓總價1.07億元。
國外情況
印度OMPL將在2013年5月底啟動芳烴項目
印度石油天然氣公司芒格洛爾石化有限公司(OMPL)將在2013年5月底啟動新芳烴項目的商業化生產。該項目包括位於新芒格洛爾的92萬噸/年對二甲苯(PX)裝置和27萬噸/年純苯裝置。
OMPL首席執行官SRamachandran參加在孟買舉辦的2012印度石化大會時表示:"該項目裝置已完成87.67%。新裝置長期出口PX和純苯的銷售招標反響不錯。"