神經母細胞瘤

神經母細胞瘤是兒童最常見的顱外腫瘤,是嬰幼兒最常見的腫瘤。有將近一半的神經母細胞瘤發生在2歲以內的嬰幼兒。神經母細胞瘤約占6-10%的兒童腫瘤,15%的兒童腫瘤死亡率。對於4歲以下兒童,每一百萬人口的死亡率為10;對於4-9歲兒童,每一百萬人口的死亡率為4例。神經母細胞瘤屬於神經內分泌性腫瘤,可以起源於交感神經系統的任意神經脊部位。其最常見的發生部位是腎上腺,但也可以發生在頸部、胸部、腹部以及盆腔的神經組織。目前已知有少數幾種人類腫瘤,可自發性地從未分化的惡性腫瘤退變為完全良性腫瘤。神經母細胞瘤就屬於其中之一。[1]
2018年12月7日,學術期刊Science發表了一項可能為治療神經母細胞瘤的新方法。
目錄
疾病簡介
不同神經母細胞瘤患者預後差別很大,也即神經母細胞瘤間存在廣泛的腫瘤異質性。根據高危因素的不同,神經母細胞瘤可以分為低危組、中危組和高危組。對於低危組神經母細胞瘤患者(最常見於嬰幼兒),靠單純的觀察或者是手術治療往往可以取得很好的效果;但是高危組神經母細胞瘤患者,即使綜合各種強化的治療方案,預後仍然不理想。嗅溝神經母細胞瘤一般被認為起源於嗅神經上皮;其分類至今尚存有爭議。因為其不屬於交感神經系統,所以嗅溝神經母細胞瘤應屬於獨立的一類腫瘤,與下文所述的神經母細胞瘤相混淆有所不同。[2]
臨床症狀
神經母細胞瘤的初發症狀不典型,因此在早期診斷有所困難。比較常見的症狀包括疲乏,食慾減退,發燒以及關節疼痛。腫瘤所導致的症狀取決於腫瘤所處的器官以及是否發生轉移。
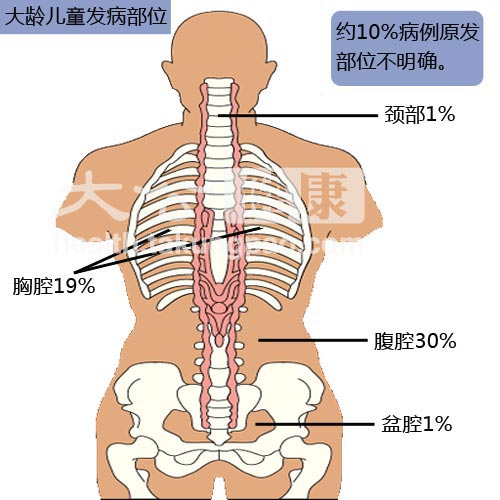
腹腔的神經母細胞瘤一般表現為腹部膨隆以及便秘;胸腔神經母細胞瘤一般表現為呼吸困難;脊髓神經母細胞瘤一般表現為軀幹與肢體力量減退,患者往往會有站立、行走等困難;腿部以及髖部等骨頭的神經母細胞瘤可以表現為骨痛以及跛行;骨髓的破壞可以使患者由於貧血所導致的皮膚蒼白。大部分神經母細胞瘤(50-60%)在出現臨床表現前,已發生廣泛轉移。原發神經母細胞瘤最常見的發生部位為腎上腺(約占40%);其他原發器官包括頸部(1%), 胸腔(19%), 腹腔(30%),以及盆腔(1%)。另有一些罕見的病例,找不到原發病灶。罕見但具有特徵性的臨床表現包括脊髓橫斷性病變(脊髓壓迫,占5%), 頑固性腹瀉(腫瘤分泌血管活性腸肽,占4%),霍納綜合徵(頸部腫瘤,占2.4%),共濟失調(腫瘤的旁分泌所致,占1.3%),以及高血壓(腎動脈受壓或者是兒茶酚胺分泌,占1.3%)。[3]





發病原因
目前關於神經母細胞瘤真正的病因尚不清楚。一些遺傳易感因素被發現與神經母細胞瘤的發病相關。家族型神經母細胞瘤被證明與間變淋巴瘤激酶(anaplastic lymphoma kinase,ALK)的體細胞突變(somatic mutation)所導致。此外,在神經母細胞瘤還發現有許多分子突變。N-myc基因的擴增突變在神經母細胞瘤也很常見。其擴增類型呈雙向分布:在一個極端為3-10倍擴增,在另一個極端為100-300倍擴增。N-myc基因的擴增突變往往與腫瘤的擴散相關。LMO1基因被證明與腫瘤的惡性程度相關。
由於神經母細胞瘤往往發生於嬰幼兒,因此有許多研究集中調查環境危險因素對懷孕以及孕期的影響。例如懷孕期間的是否接觸化學危險品、吸煙、飲酒、藥物、感染等。但是,這些研究尚未有明確的結果。
鑑別診斷
最終的診斷依賴於術後的病理,但同時也要綜合考慮患者的臨床表現以及其他的輔助檢查結果。
生化檢查
將近90%的神經母細胞瘤患者,其血液或尿液里兒茶酚胺及其代謝產物多巴胺,高香草酸、香草扁桃酸的濃度較正常人群有顯著升高。
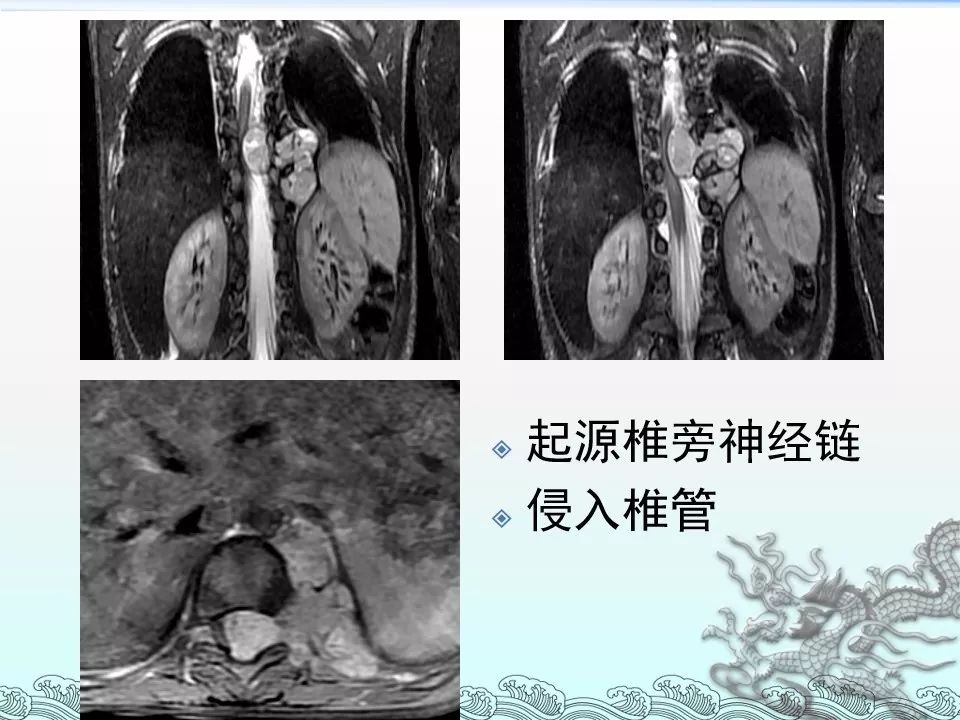
影像檢查
另外一個檢測神經母細胞瘤的手段是間位腆代苄胍(meta-iodobenzylguanidine,mIBG)掃描。該檢查的分子機理為,間位腆代苄胍是去甲腎上腺素的功能類似物(analog),並可被交感神經元所攝取。當間位腆代苄胍與放射性物質如碘-131或者是碘-123耦聯後,即可作為放射性藥物而用於神經母細胞瘤的診斷以及療效監測。碘-123的半衰期為13小時,常作為檢測的優選手段;碘-131的半衰期為8天,其在大劑量使用時,可作為治療復發以及頑固性神經母細胞瘤。[4]
免疫組化學檢查
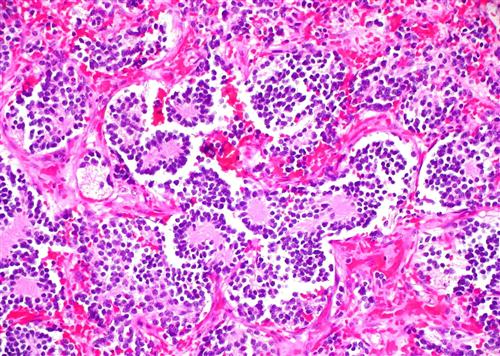
顯微鏡下,神經母細胞瘤呈現為藍染的小圓形細胞,菊花形排列。腫瘤細胞圍繞神經氈(neuropil)呈菊花形排列,與其他的腫瘤(如視神經母細胞瘤)圍繞血管呈菊花形排列有所不同。其他還有一些神經母細胞瘤所特異的免疫組化染色,用以與其他腫瘤(尤因肉瘤,淋巴瘤等)進行鑑別診斷。





腫瘤分期
國際神經母細胞瘤分期系統(International Neuroblastoma Staging System,INSS)於1986年建立並於1988年進行修訂。該系統基於腫瘤的原發器官以及轉移情況進行分期。
- 1期:局限於原發器官,無轉移灶;
- 2A期:次全切除的單側腫瘤;同側以及對側淋
巴結明確無轉移
- 2B期:次全切除或者是全切除單側腫瘤;同側淋巴結有明確轉移,而對側淋巴結明確無轉移;
- 3期:腫瘤跨中線侵襲,伴隨或未伴隨局部淋巴結轉移;或者是單側腫瘤伴有對側淋巴結轉移;或者是跨中線生長的腫瘤並伴有雙側淋巴結轉移;
- 4期:腫瘤播散到遠處淋巴結,骨髓,肝臟,或者是其他器官(除4S期所定義的器官之外)。
- 4S期:小於1歲患兒;腫瘤局限於原發器官;腫瘤擴散局限於肝臟,皮膚,或者是骨髓(腫瘤細胞少於10%的骨髓有核細胞)。
始於2005年,全球幾個主要的兒童腫瘤研究協作組,開始就1990年至2002年間在歐洲、日本、美國、加拿大以及澳大利亞地區收治的8800例神經母細胞瘤進行回顧性比對研究。根據該回顧性研究的結果,神經母細胞瘤根據風險程度的不同,進行重新分期(INRGSS)。該回顧性研究發現,12-18個月的神經母細胞瘤患兒具有良好的預後;據此,新的分類體系將不具有N-myc突變的12-18個月患兒從以前的高危組,重新劃分至中危組。該風險分類體系具體如下:
- L1期:病灶局限且無影像學確定的危險因素;
- L2期:病灶局限但具有影像學確定的危險因素;
- M期:病灶發生轉移;
- MS期:病灶發生特異性轉移(同上述的4S期);
新的風險分層體系將基於新的INRGSS分期體系、發病年齡、腫瘤級別、N-myc擴增狀態、11q染色體不均衡突變以及多核型因素,將神經母細胞瘤患者分為:極低危組、低危組、中危組以及高危組。
治療方法

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
根據上述風險分層,治療也相應有所不同:
低危組
允許進行觀察,並待疾病進展或者有變化後才進行干預;或者進行手術治療,且往往可以治癒。
中危組
手術切除並輔以化療。
高危組
大劑量化療,手術切除,放療,骨髓/造血幹細胞移植,基於13-順維甲酸的生物治療,以及基於粒細胞集落刺激生物因子與白介素2的免疫治療。
疾病預後
經治療後,低危組患者治癒率超過90%,中危組患者治癒率介於70-90%之間。然而,高危組患者的治癒率僅為30%左右。近年來,隨着免疫治療以及新藥物的出現,高危組患者的預後有了一定的提高。
科研進展
德國研究人員可能發現了一種更好的方法來治療神經母細胞瘤患者,相關研究結果發表在2018年12月7日的Science期刊上。研究人員收集了400多個神經母細胞瘤樣品,分析它們的DNA,包括尋找參與維持染色體端粒的基因突變。他們發現這些基因突變與神經母細胞瘤的侵襲性之間存在相關性。低風險的神經母細胞瘤經常缺乏此類基因突變。中度風險的神經母細胞瘤更可能具有此類基因突變。高風險的神經母細胞瘤也具有此類基因突變,但是它們在其他的關鍵基因通路(比如RAS和/或p53通路)中發生突變。該研究結果可能為治療神經母細胞瘤提供新的方法,從而為醫生提供一種可靠的方法,用於在初步診斷時確定侵襲性腫瘤發生轉移的可能性到底有多大。
視頻