皮肌炎檢視原始碼討論檢視歷史

皮肌炎(dermatomyositis,DM)又稱皮膚異色性皮肌炎(poikilodermatomyositis),屬自身免疫性結締組織疾病之一,是一種主要累及橫紋肌,呈以淋巴細胞浸潤為主的非化膿性炎症病變,可伴有或不伴有多種皮膚損害,也可伴發各種內臟損害。多發性肌炎(polymyositis,PM)系指本組疾患而無皮膚損害者。[1]
目錄
症狀體徵
根據患者對稱性近端肌肉乏力,疼痛和觸痛,伴同特徵性皮膚損害如以眶周為中心的紫紅色浮腫性斑,Gottron氏征和甲根皺襞僵直擴張性毛細血管性紅斑,一般診斷不難,再結合血清肌漿酶和CPK,LDH,AST,ALT和醛縮酶的增高,24小時尿肌酸排出量增加,必要時結合肌電圖的改變和病變肌肉的活組織檢查,可以確診本病。
可發生於任何年齡,女性略占多數,有些病例發作前有驅症狀,如不規則發熱,雷諾氏現象,關節痛,頭痛,倦急和乏力等,發病多數呈緩慢起病,少數呈急性或亞急性發病,肌肉和皮膚是本病的兩組主要症狀,皮損往往先於肌肉數周至數年發病,少數先有肌病,隨後出現皮損,部分患者肌肉和皮膚同時發病。[2]
肌肉症狀
通常累及橫紋肌,有時平滑肌和心肌亦可受累,任何部位肌肉皆可侵犯,但往往四肢肌肉首遭累及,肝體近端肌肉又比遠側的更易受損,肩胛帶和骨盆帶肌肉通常最早波及,上臂和股部肌群次之,其他部位肌群更次之,病變常呈對稱性,在少數病例中損害可局限在一個肢體肌肉群,或一單獨肌肉或許多肌肉連續發作,此起彼伏;通常患者感乏力,隨後有肌肉疼痛,按痛和運動痛;進而由於肌力下降,呈現各種運動機能障礙和特殊姿態,由於肌肉病變的多少,輕重,部位的差異等,症狀可有所不同,一般通常有抬臂,頭部運動或下蹲後站起困難,步態拙劣,有時由於肌力急遽衰減,可呈現特殊姿態,如頭部下垂,二肩前傾等,重者全身不能動彈,甚至翻身,當咽,食管上部和齶部肌肉受累時可出現聲音嘶啞和吞咽困難;當膈肌和肋間肌累及可發生氣急和呼吸困難;心肌受累可產生心力衰竭,眼肌累及發生復視,病變肌肉質地可如正常或呈柔?感,有時纖維性變後而發硬或堅實,可促使關節攣縮影響功能,亦有報導有重症肌無力病樣綜合徵即無痛性肌軟弱,在活動後加劇,病變肌肉上面的皮膚可增厚或呈水腫性。
皮膚症狀
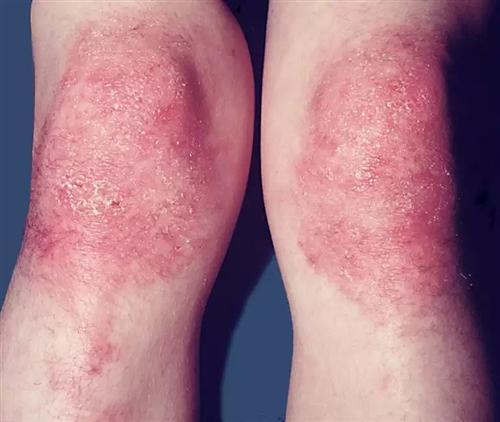
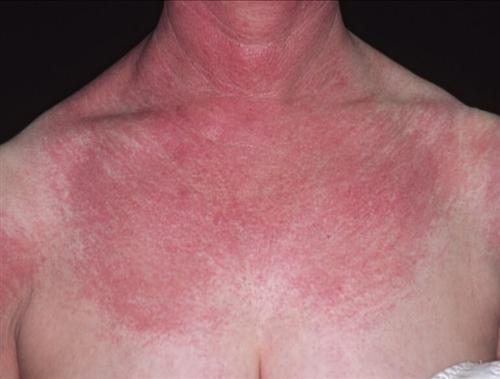
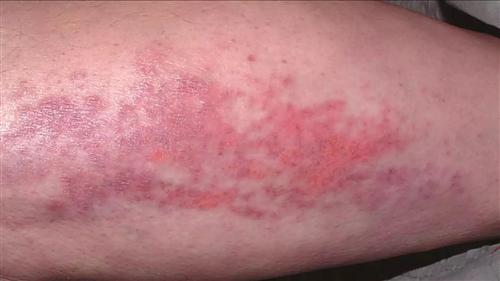
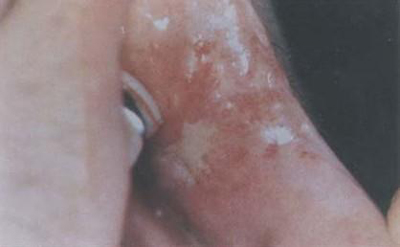
本病的皮膚損害多種多樣,有的為首發症狀;有的具有一定特異性,對診斷有幫助;有的出現提示伴發內臟惡性腫瘤;有的與預後有關,皮損病變與肌肉累及程度常不平行,有時皮損可以較為廣泛而僅有輕度肌炎,相反亦有存在嚴重肌肉病變而僅有輕度皮損,有時皮損反映了肌肉病變的程度,通常在面部特別一上眼瞼發出紫紅色斑,逐漸瀰漫地向前額,顴頰,耳前,頸和上胸部V字區等擴展,頭皮和耳後部亦可累及,閉眼近瞼緣處可見明顯擴張的樹枝狀毛細血管,偶見彎曲頂端有針頭大小瘀點的毛血管;以眼瞼為中心出現眶周不等程度浮腫紫紅色斑片具有一定特徵性,四肢肘膝尤其掌指關節和指間關節伸面出現紫紅色丘疹,斑,以後變萎縮,有毛細血管擴張,色素減退和上覆細小鱗屑,偶見潰破,稱Gottron征,亦具有特徵性,在甲根皺襞可見僵直毛細血管擴張和瘀點,有助於診斷,有些病例軀幹部亦可出現皮疹,呈瀰漫性或局限性暗紅色斑或丘疹,位在胸骨前或肩胛肌間或腰背部皮膚,通常皆無瘙癢,疼痛,感覺異常,但少數病例可有劇癢,損害呈暫時性,反覆發作,其後相互融合,持續不退,上有細小鱗屑,口腔粘膜亦出現紅斑。[3]
在慢性病例中有時尚可出現多發角化性小丘疹,斑點狀色素沉着,行細血管擴張,輕度皮膚萎縮和色素脫失,稱血管萎縮性異色病性皮肌炎,偶而在異色病樣疹基礎上皮疹呈現炎紅色甚至棕紅色,損害廣泛,尤以頭面部為著,象酒醉後外觀,伴較多深褐色,灰褐針頭大色素斑,並可見林量蟠曲樹枝狀成堆成團擴張的毛細血管,稱之惡性紅斑,常提示伴發惡性腫瘤。
此外可有皮下結節,鈣質沉積排出皮膚形成漏管,有時在非典型病例中僅在眼瞼,一側或兩側或鼻根部出現紫紅色斑,或頭皮部出現瀰漫性性紅斑,糠枇樣脫屑,脫髮,或為蕁麻疹,多形紅斑樣,網狀青斑,雷諾現象等,部分病例對日光過敏。
近年來文獻中報告約有8%病例,只有皮疹,經長期隨訪亦未見肌肉病變稱皮膚型皮肌炎。
小兒患者除上描述外,其特點是發病前常有上呼吸感染史,無雷諾現象和硬皮病樣變化,在皮膚,肌肉,筋膜中有瀰漫或局限性發生鈣質沉着,較成人為常見,有血管病變,腸胃道出現潰瘍和出血,與成人不同。
此外,患者可膛規則發熱,發熱可為本病的初發症狀,亦可在本病的發展過程中發生,常為不規則低熱,在急性病例中熱度可較高,約40%病例有發熱;可有關節痛,肘膝肩和指關節發生畸形和運動受阻,多數繼發於鄰近肌肉病變的纖維經攣縮所致,X線攝片在有些病例中見關節間隙消失,骨皮質破壞,約20%有關節病變,淺表淋巴結一般無明顯腫大,少數頸部淋巴結可成串腫大;心臟累及病例有心功能異常,心動過速或過緩,心臟擴大,心肌損害,房顫和心力衰竭,亦可有胸膜炎,間質性肺炎,約1/3病例肝輕度至中等度腫大,質中堅,消化道累及鋇餐示食管蠕動差,通過緩慢,食管擴張,梨狀鋇劑滯留,眼肌累及呈復視,視網膜有時有滲出物或出血,或有視網膜脈絡膜炎,蛛網膜下腔出血。
此外,本病可與SLE和硬皮病等病重疊。






治療方法
在無腫瘤並發的病例,皮質類固醇治療有效,一般成人劑量相等於潑尼松60~100mg/d,約為1mg/(kg·d),重症病例或開始劑量無效,可增至1.5mg/(kg·d);兒童劑量通常較成人劑量增加些,為1.5mg/(kg·d)。症狀輕者可用較小劑量。根據臨床症狀,尿肌酸排出量和血清肌漿酶測定值作為應用皮質炎固醇增減劑量的參考指標,一般肌力恢復較肌漿酶和尿肌酸排泄量好轉遲緩數周。近年來重症病例採用大劑量甲基潑尼松龍衝擊療法(即靜脈滴注1g,連續3天,以後再改用潑尼松600mg/d)。約1/3病例對皮質類固醇治療效應不佳。
免疫抑制劑特別是氨甲喋呤靜脈滴注合併皮質類固醇治療尤其對改善肌力有一定療效,環磷酰胺和硫唑嘌呤也可應用。其他非甾體類抗體炎藥物,蛋白同化激素如苯丙酸諾龍、抗瘧藥物(如氯化喹啉)和維生素E等亦可輔助試用。重症病例可靜脈補給複方氨基酸注射液,三磷酸腺苷、輔酶A和能量合劑。近亦有應用環胞菌素、血漿透析等獲得一定效果。此外物理療法,在急性期嚴重炎症時進行被動運動防止軟攣縮,每日二次,不鼓勵主動運動;在恢復期鼓勵進行速度緩慢主動運動。其他可酌情採用按摩、推拿水療,透熱電療等以防止肌肉萎縮和攣縮。對功能消失患者進行康復治療訓練。[4]
在成人特別是40~50歲以上患者,必需詳細地檢查有無腫瘤的伴發,如果發現腫瘤需予以徹底治療,可改善和緩解皮肌炎症狀。如果當時未發現,亦應每隔3~6個月定期隨訪甚為必要。對小兒皮肌炎患者,需儘量去除一切可疑病灶,並採用抗生素合併皮質類固醇治療,可獲良效。
飲食保健

根據不同的症狀,有不同情況的飲食要求,具體詢問醫生,針對具體的病症制定不同的飲食標準。
預防護理
本病病程大部分病例為慢性漸進性,在2~3年趨向逐步恢復,僅少數死亡,故少數發作急性呈顯著乏力的病例,多數預後不良,常由於並發感染死亡,另有小部分病例呈反覆發作,加劇與緩解交替進行,最終獲得緩解。
本病並發腫瘤的百分數從9%至52%不等,一般在40歲以後,發病年齡愈大,伴發腫瘤的機會越大,有報導在50歲以上男性患者中可高達71%,Schuerman複習文獻的344例,12%伴發惡性腫瘤,William報告為15%,Gallen為24%,皮肌炎患者伴發惡性腫瘤的發生率遠超過多發性肌炎患者伴發的,有人認為與皮肌炎患者應用免疫抑制劑後有關,作者報導的135例中有12例(8.89%)伴發,大多先有皮肌炎,隨後發生腫瘤,發生腫瘤的部位依次為胃,卵巢,子宮,膽囊,鼻咽,肺,食管等。
病理病因
確切病因尚不夠清楚,可能為病毒感染,機體免疫異常對自我的異常識別以及血管病變,三者亦可能有相互聯繫,例如橫紋肌纖維的慢病毒感染可導致肌纖維抗原性的改變,被免疫系統誤認為「異已」,從而產生血管炎而發生本病。
- 免疫學研究鑑於患者血清免疫球蛋白增高,肌肉活檢標本示微小血管內有IgG,IgM和C3以及補體膜攻擊複合物C56-C9沉積,沉着的程度似與疾病活動性相關,Arahata和Engel證實在DM的炎症性病灶中有B細胞的顯著增多,提示局部體液效應的增強,但亦有學者認為這些抗體的沉積是肌肉損傷的後果而非其原因,亦有學者發現患者周圍血淋巴細胞在加入橫紋肌抗原後其轉化率以及巨噬細胞移動抑制試驗對照組為高,且與其活動度呈正相關,經用糖皮質激素減低,患者周圍血淋巴細胞在體外組織培養對肌母細胞有細胞毒作用,其損傷作用可能是釋放淋巴毒素或直接粘附和侵入肌纖維。有人認本病與SLE和硬皮病等有許多共同的臨床和免疫學異常,如部分病例可找到LE細胞,抗核抗體和類風濕因子檢測陽性,用熒光抗體技術在表皮基底膜,血管壁可見免疫球蛋白沉積,且血清中發現有抗多發性肌炎抗原-1(簡稱抗PM-1)和抗肌凝蛋白抗體,故提出自身免疫疾病學說,又如在伴發惡性腫瘤患者,腫瘤的切除可使本病症狀緩解,用患者腫瘤提出液做皮內試驗呈現陽性反應,且被動轉移試驗亦為陽性,患者血清中發現有對腫瘤的抗體,這些惡性腫瘤作為機體自身抗原而引起抗體的產生,又腫瘤組織可與體內正常的肌纖維,腱鞘,血管,結締組織間發生交叉抗原性,因而能與產生的抗體發生交叉的抗原抗體反應,導致這些組織的病變,從而作為本病自身免疫患學說的依據。
- 感覺學說近年來有學者將患者的肌肉和皮損作電鏡觀察,發現肌細胞核內,血管內皮細胞,血管周圍的組織細胞和成纖維細胞漿和核膜內有類似粘病毒或副粘病毒的顆粒,近報告從11歲女孩病變肌肉中分離出柯薩奇(Coxackie)A9病毒,故提出感染學說,然而在動物實驗中至今未能在注射患者的肌肉,血漿而導致肌肉炎症,從患者血液中不能測出抗病毒的抗體。在小兒皮肌炎患者,發病前常有上呼吸道感染史,抗鏈球菌「O」值增高,以抗生素合併皮質固醇治療可獲良效,提出感染變態反應學說。
- 血管病變學說血管病變特別在兒童型DM曾被描述,任何瀰漫性血管病變可以產生橫紋肌的缺血,從而引起單個纖維的壞死和肌肉的梗死區,在DM/PM特別兒童患者中有毛細血管的內皮細胞損傷和血栓的證據,且有免疫複合物沉積在肌肉內血管中,以及毛細血管基底膜增厚,毛細血管減少特別在肌束周區。






疾病診斷
本病需與下列疾病相鑑別:
- 系統性紅斑狼瘡皮損以顴頰部水腫性蝶形紅斑,指(趾)節伸面暗紅斑和甲周為中心的浮腫性紫紅斑,指(趾)間關節和掌(?)指(趾)關節伸面紫紅斑以及甲根皺襞的僵直毛細血管擴張紅斑有所區別;SLE多系統病變中以腎主要累及而皮肌炎以肢體近端肌肉累及為主,聲音嘶啞和吞噬困難亦較常見,此外血清肌漿酶和尿肌酸排出量的測定在皮肌炎患者有明顯增高,需要時肌電圖和肌肉活組織檢查可資鑑別。
- 系統性硬皮病;皮肌炎的後期病變如皮膚硬化,皮下脂肪組織中鈣質沉着,組織學上也可見結締組織腫脹,硬化,皮膚附近萎縮等,但在系統性硬皮病病初期,有雷諾氏現象,顏面和四肢末端腫脹,硬化以後萎縮為其特徵,肌肉病變方麵皮肌炎初期病變即已顯著,為實質性肌炎,而在系統性硬皮病中肌肉病變通常在晚期出現,且為間質性肌炎可作鑑別。
- 風濕性多肌痛症(polymyalgia rheumatica),通常發生在40歲以上,上肢近端發生瀰漫性疼痛較下肢為多,伴同全身乏力,患者不能道出來疼痛來自肌肉還是關節,無肌無力,由於失用可有輕度消瘦,血清CPK值正常,肌電圖正常或輕度肌病性變化。
- 嗜酸性肌炎(eosinophilic myositis) 其特徵為亞急性發作肌痛和近端肌群無力,血清肌漿酶可增高,肌電圖示肌病變化,肌肉活檢示肌炎伴同嗜酸性細胞炎性浸潤,有時呈局灶性變化,為嗜酸性細胞增多綜合徵病譜中的一個亞型。
檢查方法
血象通常無顯著變化,有時有輕度貧血和白細胞增多,約1/3病例有嗜酸性粒細胞增高,紅細胞沉降率中等度增加,血清蛋白總量不變或減低,白球蛋白比值下降,白蛋白減少,α2和γ球蛋白增加。
免疫學檢測
DM/PM患者血清中可檢測出兩類自身抗體。
- 直接抗肌肉及其成份的抗體 Wada等用高度純化的肌漿球蛋白經放射免疫測定,發現PM患者的血清中肌漿球蛋白抗體的陽性率為90%,其他結締組織病患者未發現此抗體,Nishikai等發現肌炎中患者的肌紅蛋白抗體的陽性率為71%,其他結締組織病患者低於15%,正常人則未發現。
- 抗核抗體和細胞漿抗體 LE細胞約10%陽性,抗核抗體約1/5~1/3病例陽性,核型主要為微小斑點型。
- 抗J0-1抗體:抗原為組胺酰tRNA全成酶,抗胞漿抗體,PM中陽性率為30%~40%,DM中<5%,兒童型DM中罕見,亦可見於重疊綜合徵尤其伴有乾燥綜合徵患者,其間質性肺部疾患密切關聯。
- 抗Mi-2抗體:Mi-2抗原為一核蛋白,約8%病例陽性,兒童型DM及伴惡性腫瘤的DM偶見。
- 抗PM-1/PM-Scl抗體:抗原為核仁蛋白,陽性率為8~12%,亦可見於與硬皮病重疊的病例。
- 抗PL-7抗體:即抗蘇氨酰tRNA合成酶抗體,肌炎患者中陽性率為3%~4%。
- 抗PL-12抗體:即抗丙氨酰tRNA合成酶抗體,陽性率為3%,在非肌炎患者中抗PL-7和PL-12抗體均屬罕見,兩者與J0-1抗體相關的疾患為同一亞類肌炎。
- 肌炎患者中發現的其他細胞漿抗體有:
- Fer抗體;
- Mas抗體,兩者均屬罕見;
- Ro/SS-A抗體和La/ss-B抗體,肌炎患者的陽性率通常為7%~8%,常見於與其他結締組織病重疊的病例;
- 抗U1SmRNP抗體,在DM/PM患者中陽性率為10%~15%。
3.其他免疫學檢查 約1/3患者C4輕度至中等度降低,C3偶而減少,有報告DM瘵遺傳性C2缺陷,有的病例CIC增高。直接免疫熒光法測定病變肌肉中毛細胞血管壁特別是兒童病例顯示有IgG,IgM和補體沉積,但在病變皮膚皮損局灶性真皮表皮交界處可見局灶性Ig和C沉積,但無連續性沉積,與SLE不同。
尿肌酸排泄量增加

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
正常情況肌酸的合成過程首先是精氨酸將脒基轉移給甘氨酸而成胍乙酸,其次是胍乙酸接受蛋氨酸的甲基成為肌酸,在肝臟內合成,大部分由肌肉攝取,以含高能磷酸鍵的磷酸肌酸形式存在,其中高能磷酸鍵在磷酸肌酸激酶的催化下可轉移給二磷酸腺苷而生成三磷酸腺苷,當三磷酸腺苷合成增加時,一部分磷酸即可通過逆向反應而儲存在磷酸肌酸中,肌酸在肌肉內代謝脫水形成肌酐以後從尿中排出,在發育期,婦女月經來潮前後和老年人可有生理性肌酸尿,但其24小時排出總量不超過每千克體重4mg,患本病時由於肌肉的病變,所攝取的肌酸減少,參加肌肉代謝活動的肌酸量亦減少,形成肌酐量因之亦減少,血中肌酸量增高而肌酐量降低,肌酸從尿中大量排出而肌酐排出量卻降低,皮肌炎患者24小時肌酸排出量甚至可高達2g。
血清肌漿酶測定
血清肌酸磷酸激酶(CPK),醛縮酶,(AST),丙氨酸氨基轉移酶(ALT),乳酸脫氫酶測定值增高,特別CPK,血清酶的增高常與本病肌肉病變的消長平行,可反映疾病的活動性,一般在肌力改善前3~4周降低,臨床復發前5~6周升高,可預示病情的惡化,當患者CPK值增高,其CKMM1:CKMM<30%和CKMM1:>1時提示PM病情嚴重,未經治療的活動性肌炎患者通常異常,但此酶對肌肉無特異性,肝臟內具有較多的醛縮酶,肝病時該酶亦可增高,碳酸酐酶Ⅲ為唯一在於骨骼肌的同工酶,骨骼肌損傷時可增高。
肌電圖改變
呈肌原性萎縮相,常見的為失神經纖維性顫動,病變肌肉示失神經現象,呈現不規則不隨意的放電波形,罹患肌肉不是全部肌纖維同樣受累,其中多半有正常的肌纖維散在,輕用力時呈短時限的多相運動單位,最大用力時呈低電壓干擾相多波增加。
組織學改變
- 肌肉改變 肌肉廣泛或部分地受侵犯,肌纖維初期呈腫脹,橫紋消失,肌漿透明化,肌纖維膜細胞核增加,肌纖維分離,斷裂,在進行性病變中肌纖維可呈玻璃樣,顆粒狀,空泡狀等變性,有時甚至壞死,或肌肉結構完全消失代以結締組織,有時可見鈣質沉着,間質示炎症性改變,血管擴張,內膜增厚,管腔狹窄,甚至栓塞,血管周圍有淋巴細胞伴同漿細胞和組織細胞浸潤,主要發生在橫紋肌中,有些病例平滑和心肌也可發生相同病變。
- 亦有學者認為DM最特徵性的病理改變為束周萎縮,即肌纖維的萎縮和損傷常集中於肌束周圍,橫斷面上往往見肌束邊緣的肌纖維直徑明顯縮小,產生原因有人認為DM的病變較多局限於肌外衣,肌外衣的纖維性增厚造成環繞肌束邊緣的肌纖維萎縮,亦有人解釋束周萎縮是血管損傷處外周肌束血管中斷造成慢性缺血的結果。
- 皮膚改變 在初期水腫性紅斑階段,可見表皮角化,棘層萎縮,釘突消失,基底細胞液化變性,真皮全層粘液性水腫,血管擴張,周圍為主淋巴細胞浸潤,間有少許組織細胞,有色素失禁,在進行性病變中,膠原纖維腫脹,均質化和硬化,血管壁增厚,皮下脂肪組織粘液樣變性,鈣質沉着,表皮進一步萎縮,皮膚附什亦萎縮。
其他
肌紅蛋白存在於骨骼和心肌中,政黨人血和尿中僅有少量肌紅蛋白,嚴重的肌損傷可釋放大量的肌紅蛋白,血清肌紅蛋白測定可作為衡量疾病活動程度的指標,尿中出現可見的血紅蛋白樣色素;病情加重時排出增多,緩解時減少,亦有報導尿3-甲基組氨酸排出增多為肌肉損傷的標誌,缺點是無特異性。
併發症
可有關節痛,肘膝肩和指關節發生畸形和運動受阻,多數繼發於鄰近肌肉病變的纖維經攣縮所致,心臟累及病例有心功能異常,心動過速或過緩,心臟擴大,心肌損害,房顫和心力衰竭,亦可有胸膜炎,間質性肺炎,約,/,病例肝輕度至中等度腫大,質中堅,消化道累及鋇餐示食管蠕動差,通過緩慢,食管擴張,梨狀鋇劑滯留,眼肌累及呈復視,視網膜有時有滲出物或出血,或有視網膜脈絡膜炎,蛛網膜下腔出血。
視頻
視頻