多發性骨髓瘤

多發性骨髓瘤(multiple myeloma,MM)是惡性漿細胞病中最常見的一種類型,又稱骨髓瘤、漿細胞骨髓瘤或Kahler病。雖然早在1844年對此病已有人作出描述,但直到1889年經Kahler詳細報告病例後,多發性骨髓瘤才普遍為人們所了解和承認。多發性骨髓瘤的特徵是單克隆漿細胞惡性增殖並分泌大量單克隆免疫球蛋白。惡性漿細胞無節制地增生、廣泛浸潤和大量單克隆免疫球蛋白的出現及沉積,正常多克隆漿細胞增生和多克隆免疫球蛋白分泌受到抑制,從而引起廣泛骨質破壞、反覆感染、貧血、高鈣血症、高黏滯綜合徵、腎功能不全等一系列臨床表現並導致不良後果。[1]
目錄
症狀體徵
多發性骨髓瘤臨床表現多種多樣,有時患者的首發症狀並不引人直接考慮到本病的可能,若不警惕本病並作進一步檢查,則易發生誤診或漏診。
- 骨痛:骨痛是本病的主要症狀之一。疼痛程度輕重不一,早期常是輕度的、暫時的,隨着病程進展可以變為持續而嚴重。疼痛劇烈或突然加劇,常提示發生了病理性骨折。據北京協和醫院125例MM首發症狀分析,80例(64.0%)以骨痛為主訴,骨痛部位以腰骶部最常見(28.0%),其次為胸肋骨(27.0%),四肢長骨較少(9.0%),少數患者有肩關節或四肢關節痛。絕大多數(90%~93%)患者在全病程中都會有不同程度的骨痛症狀,但確有少數患者始終無骨痛。除骨痛、病理骨折外,還可出現骨骼腫物,瘤細胞自骨髓向外浸潤,侵及骨皮質、骨膜及鄰近組織,形成腫塊。在多發性骨髓瘤,這種骨骼腫塊常為多發性,常見部位是胸肋骨、鎖骨、頭顱骨、鼻骨、下頜骨及其他部位。與孤立性漿細胞瘤不同的是,其病變不僅是多發的,而且骨髓早已受侵犯,並有大量單克隆免疫球蛋白的分泌。
- 貧血及出血傾向:貧血是本病另一常見臨床表現。據北京協和醫院125例分析,絕大多數(90%)患者都在病程中出現程度不一的貧血,其中部分(10.4%)患者是以貧血症狀為主訴而就診。貧血程度不一,一般病程早期較輕、晚期較重,血紅蛋白可降到<50g/L。造成貧血的主要原因是骨髓中瘤細胞惡性增生、浸潤,排擠了造血組織,影響了造血功能。此外,腎功不全、反覆感染、營養不良等因素也會造成或加重貧血。出血傾向在本病也不少見。北京協和醫院125例中8例是以出血為首發症狀而就醫,而在病程中出現出血傾向者可達10%~25%。出血程度一般不嚴重,多表現為黏膜滲血和皮膚紫癜,常見部位為鼻腔、牙齦、皮膚,晚期可能發生內臟出血及顱內出血。導致出血的原因是血小板減少和凝血障礙。血小板減少是因骨髓造血功能受抑,凝血障礙則因大量單克隆免疫球蛋白覆蓋於血小板表面及凝血因子(纖維蛋白原,凝血酶原,因子Ⅴ、Ⅶ、Ⅷ等)表面,影響其功能,造成凝血障礙。免疫球蛋白異常增多使血液黏度增加,血流緩慢不暢,損害毛細血管,也可造成或加重出血。[2]
- 反覆感染:本病患者易發生感染,尤以肺炎球菌性肺炎多見,其次是泌尿系感染和敗血症。病毒感染中以帶狀皰疹、周身性水痘為多見。北京協和醫院125例中以發熱、感染為主訴而就醫者18例(占14.4%),其中多數系肺部感染。部分患者因反覆發生肺炎住院,進一步檢查方確診為MM並發肺炎。對晚期MM患者而言,感染是重要致死原因之一。本病易感染的原因是正常多克隆B細胞——漿細胞的增生、分化、成熟受到抑制,正常多克隆免疫球蛋白生成減少,而異常單克隆免疫球蛋白缺乏免疫活性,致使機體免疫力減低,致病菌乘虛而入。此外,T細胞和B細胞數量及功能異常,以及化療藥物和腎上腺皮質激素的應用,也增加了發生感染的機會。
- 腎臟損害:腎臟病變是本病比較常見而又具特徵性的臨床表現。由於異常單克隆免疫球蛋白過量生成和重鏈與輕鏈的合成失去平衡,過多的輕鏈生成,相對分子質量僅有23000的輕鏈可自腎小球濾過,被腎小管重吸收,過多的輕鏈重吸收造成腎小管損害。此外,高鈣血症、高尿酸血症、高黏滯綜合徵、澱粉樣變性及腫瘤細胞浸潤,均可造成腎臟損害。患者可有蛋白尿、本-周(Bence- Jones)蛋白尿、鏡下血尿,易被誤診為「腎炎」。最終發展為腎功能不全。腎功能衰竭是MM的致死原因之一。在大多數情況下,腎功能衰竭是慢性、漸進性的,但少數情況下可發生急性腎功能衰竭,主要誘因是高鈣血症和脫水,若處理及時得當,這種急性腎功能衰竭還可逆轉。
- 高鈣血症:血鈣升高是由於骨質破壞使血鈣逸向血中、腎小管對鈣外分泌減少及單克隆免疫球蛋白與鈣結合的結果。增多的血鈣主要是結合鈣而非離子鈣。血鈣>2.58mmol/L即為高鈣血症。高鈣血症的發生率報告不一,歐美國家MM患者在診斷時高鈣血症的發生率為10%~30%,當病情進展時可達30%~60%。我國MM患者高鈣血症的發生率約為16%,低於西方國家。高鈣血症可引起頭痛、嘔吐、多尿、便秘,重者可致心律失常、昏迷甚至死亡。鈣沉積在腎臟造成腎臟損害,重者可引起急性腎功能衰竭,威脅生命,故需緊急處理。
- 高黏滯綜合徵:血中單克隆免疫球蛋白異常增多,一則包裹紅細胞,減低紅細胞表面負電荷之間的排斥力而導致紅細胞發生聚集,二則使血液黏度尤其血清黏度增加,血流不暢,造成微循環障礙,引起一系列臨床表現稱為高黏滯綜合徵。常見症狀有頭暈、頭痛、眼花、視力障礙、肢體麻木、腎功能不全,嚴重影響腦血流循環時可導致意識障礙、癲癇樣發作,甚至昏迷。眼底檢查可見視網膜靜脈擴張呈結袋狀擴張似「香腸」,伴有滲血、出血。因免疫球蛋白包裹血小板及凝血因子表面,影響其功能,加之血流滯緩損傷毛細血管壁,故常有出血傾向,尤以黏膜滲血(鼻腔、口腔、胃腸道黏膜)多見。在老年患者,血液黏度增加、貧血、血容量擴增可導致充血性心力衰竭發生。雷諾現象也可發生。高黏滯綜合徵的發生既與血中免疫球蛋白濃度有關,也與免疫球蛋白類型有關。當血液黏度(血漿或血清黏度)超過正常3倍以上、血中單克隆免疫球蛋白濃度超過30g/L時,易發生高黏滯綜合徵。在各種免疫球蛋白類型中,IgM相對分子質量大、形狀不對稱,並有聚集傾向,故最易引起高黏滯綜合徵。其次,IgA和IgG3易形成多聚體,故也較易引起高黏滯綜合徵。
- 高尿酸血症:血尿酸升高>327μmol/L者在MM常見。北京協和醫院MM 91例中,61例(67%)有高尿酸血症。血尿酸升高是由於瘤細胞分解產生尿酸增多和腎臟排泄尿酸減少的結果。血尿酸升高雖然很少引起明顯臨床症狀,但可造成腎臟損害,應予預防和處理。
- 神經系統損害:瘤細胞浸潤、瘤塊壓迫、高鈣血症、高黏滯綜合徵、澱粉樣變性以及病理性骨折造成的機械性壓迫,均可成為引起神經系統病變和症狀的原因。神經系統症狀多種多樣,既可表現為周圍神經病和神經根綜合徵,也可表現為中樞神經系統症狀。胸椎、腰椎的壓縮性病理性骨折可造成截癱。北京協和醫院125例中12例有神經系統病變,周圍神經病變3例、神經根損害3例、顱內損害2例、脊髓受壓而致截癱4例。
- 澱粉樣變性:免疫球蛋白的輕鏈與多糖的複合物沉澱於組織器官中即是本病的澱粉樣變性。受累的組織器官常較廣泛,舌、腮腺、皮膚、心肌、胃腸道、周圍神經、肝、脾、腎、腎上腺、肺等均可被累及,可引起舌肥大、腮腺腫大、皮膚腫塊或苔蘚病、心肌肥厚、心臟擴大、腹瀉或便秘、外周神經病、肝脾腫大、腎功能不全,等等。澱粉樣變性的診斷依賴組織活檢病理學檢查,包括形態學、剛果紅染色及免疫熒光檢查。歐美國家報告澱粉樣變性在MM的發生率為10%~15%,而我國的發生率為1.6%~5.6%。由澱粉樣變性損害正中神經引起的「腕管綜合徵」(carpal tunnel syndrome)在西方國家多見,而國內尚未見有報告。
- 肝脾腫大及其他:瘤細胞浸潤、澱粉樣變性導致肝脾腫大。肝大見於半數以上患者,脾大見於約20%患者,一般為肝、脾輕度腫大。淋巴結一般不腫大。少數患者可有關節疼痛,甚至出現關節腫脹、類風濕樣結節,系骨關節發生澱粉樣變性的表現。皮膚損害如瘙癢、紅斑、壞疽樣膿皮病、多毛僅見於少數患者。個別患者有黃瘤病,據認為是單克隆免疫球蛋白與脂蛋白結合的結果。[3]
併發症
- 骨折:病理性骨折,常見於顱骨、盆骨、肋骨、脊柱骨骨折等。
- 高鈣血症:骨髓瘤合併高鈣血症在歐美患者中的發生率可達30%~60%,臨床可表現為食欲不振、噁心、嘔吐、煩渴性多尿、昏迷。
- 腎臟損害:是MM常見和重要的併發症,也是患者死亡的主要原因之一。
- 高黏滯綜合徵:在MM患者中發生率為10%,常表現視力下降、意識障礙、中樞神經系統紊亂、心衰等。
- 血液系統併發症:貧血、出血、血栓。
- 感染:在病程中可反覆出現感染、發熱。如皮膚感染、肺部感染等。
- 澱粉樣變性:引起相應的臨床表現,包括舌肥大、腮腺腫大、心肌肥厚、心臟擴大、腹瀉、外周神經病、肝脾腫大等。
- 神經系統損害:MM合併神經系統損害的發病率28.6%~40%,包括脊髓壓迫、神經根脊髓壓迫等。







病理病因
MM的病因迄今尚未完全明確。臨床觀察、流行病學調查和動物實驗提示,電離輻射、慢性抗原刺激、遺傳因素、病毒感染、基因突變可能與MM的發病有關。MM在遭受原子彈爆炸影響的人群和在職業性接受或治療性接受放射線人群的發病率顯著高於正常,而且接受射線劑量愈高,發病率也愈高,提示電離輻射可誘發本病,其潛伏期較長,有時長達15年以上。據報告化學物質如石棉、砷、殺蟲劑、石油化學產品、塑料及橡膠類的長期接觸可能誘發本病,但此類報告大都比較零散,尚缺乏足夠令人信服的證據。臨床觀察到患有慢性骨髓炎、膽囊炎、膿皮病等慢性炎症的患者較易發生MM。動物試驗(向小鼠腹腔注射礦物油或包埋塑料)證明慢性炎症刺激可誘發腹腔漿細胞瘤。MM在某些種族(如黑色人種)的發病率高於其他種族,居住在同一地區的不同種族的發病率也有不同,以及某些家族的發病率顯著高於正常人群,這些均提示MM的發病可能與遺傳因素有關。病毒與MM發病有關已在多種動物試驗中得到證實,早先有報告EB病毒與人多發性骨髓瘤發病有關,近年來又報道Human Herpes Virus-8(HHV-8)與MM發病有關。但是究竟是偶合抑或是病毒確與MM發病有關,尚待進一步研究澄清。MM可能有多種染色體畸變及癌基因激活,但未發現特異的標誌性的染色體異常。染色體畸變是否是MM發病的始動因素,尚待研究證實。惡性腫瘤是多因素、多基因、多步驟改變導致的疾病,MM也不例外。[4]
疾病診斷
多發性骨髓瘤是較易發生誤診的內科疾患之一。在臨床上常被誤診為骨質疏鬆、骨轉移癌、腰椎結核、腎病、復發性肺炎、泌尿系感染等病。在診斷時又需與反應性漿細胞增多症、意義未明單克隆免疫球蛋白血症、原發性巨球蛋白血症、原發性系統性澱粉樣變性、伴發於非漿細胞病的單克隆免疫球蛋白增多、骨轉移癌、原發於骨的腫瘤、原發性腎病、甲狀旁腺功能亢進等病鑑別。國內曾有報道2547例MM的臨床誤診率高達69%,可見MM的鑑別診斷是臨床醫師應該注意的重要問題。
1.反應性漿細胞增多症:因此,多種病原體病毒、結核菌等、抗原藥物、腫瘤等、機體免疫功能紊亂舍格倫綜合徵、類風濕性關節炎等均可引起反應性漿細胞增多和免疫球蛋白水平增高,需與多發性骨髓瘤鑑別。鑑別要點如下:
- 骨髓瘤中漿細胞增多有限:一般≥3%但<10%且均為正常成熟漿細胞,而MM骨髓漿細胞常>15%且有幼稚漿細胞骨髓瘤細胞出現。
- 反應性漿細胞增多症:所分泌的免疫球蛋白屬正常多克隆性且水平升高有限(如IgG<30g/L),而MM分泌的免疫球蛋白是單克隆性(即M成分)且水平升高顯著(如IgG>30g/L)。
- 反應性漿細胞增多症本身不引起臨床症狀:其臨床表現取決於原發病,故無貧血、骨痛、骨質破壞、低白蛋白血症、正常免疫球蛋白減少、高鈣血症、高黏滯綜合徵等MM的相關臨床表現。
- 反應性漿細胞增多症有其原發性疾病的臨床表現。
2.意義未明單克隆免疫球蛋白血症MGUS MGUS和MM同為老年性疾患,且都有單克隆免疫球蛋白增多,兩者有相似之處,易於混淆。但是,MGUS不需治療,僅需隨診觀察,而MM為惡性腫瘤,應接受治療,且預後不良,故需注意兩者的鑑別(表4)。
應當強調,在符合MGUS診斷標準的患者中,有相當部分患者最終會發展為MM或其他惡性漿細胞病或B淋巴細胞惡性疾病。Kyle等報告,1960~1999年在Mayo臨床醫學中心診斷MGUS 1384例,長期隨診10年後12%、隨診20年後20%、隨診30年後25%的MGUS將發展為MM或其他漿細胞疾病(巨球蛋白血症、系統性澱粉樣變性)或B淋巴細胞惡性增殖性疾病(慢性淋巴細胞白血病、非霍奇金淋巴瘤),即MGUS以每年1%的速度轉化為惡性疾病,其中主要是轉化為MM。Cesana等報告1104例MGUS,隨診中位時間65個月(12~239個月),64例(5.8%)發展為MM,1例發展為髓外漿細胞瘤,12例發展為Waldenstrom巨球蛋白血症,6例發展為非霍奇金淋巴瘤,1例發展為慢性淋巴細胞白血病。Gregerson等報告丹麥的North Jutland在1978~1993年共診斷MGSU 1324例。其中97例(9.3%)最終發展為MM或其他惡性漿細胞病。
MGUS轉化為MM或其他惡性漿細胞病的機制尚未闡明。Rasillo和Konigsherg等的研究提示,染色體13q-與MGUS轉化為MM'
檢查方法
實驗室檢查
實驗室檢查對MM的診斷、分型、臨床分期及預後判斷都有重要意義。
- 外周血:貧血見於絕大多數患者,隨病情進展而加重。一般屬正細胞正色素性貧血,但可有大細胞性貧血伴骨髓幼紅細胞巨幼樣變,也可因有失血而表現小細胞低色素性貧血。紅細胞常呈緡錢狀排列,血沉也明顯加快,常達80~100mm/h以上,此因異常球蛋白包裹紅細胞表面使紅細胞表面負電荷之間排斥力下降而相互聚集的結果。紅細胞聚集現象可能給紅細胞計數、血型檢查造成困難。白細胞計數正常或減少。白細胞減少與骨髓造血功能受損及白細胞凝集素的存在有關。白細胞分類計數常顯示淋巴細胞相對增多至40%~55%。外周血塗片偶可見到個別瘤細胞,若出現大量瘤細胞,應考慮為漿細胞白血病。血小板計數正常或減少。血小板減少的原因是骨髓造血功能受抑和血小板凝集素存在的緣故。當血小板表面被異常球蛋白覆蓋時,功能受到影響,可成為出血的原因之一。
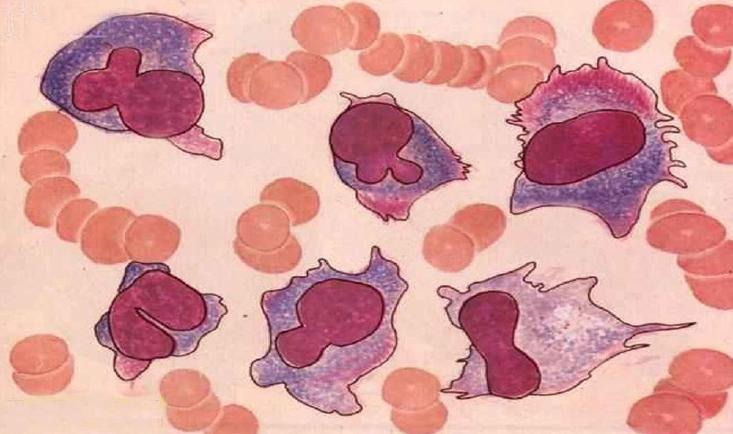
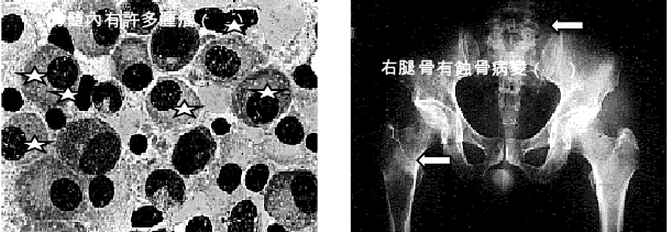
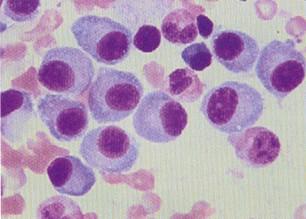
- 骨髓象:骨髓瘤細胞的出現是MM的主要特徵。瘤細胞數量多少不等,一般都占有核細胞5%以上,多者可達80%~95%以上。骨髓一般呈增生性骨髓象,各系統比例與瘤細胞數量有關,當瘤細胞所占比例較小時,粒細胞和紅細胞系比例可大致正常,巨核細胞數也可在正常範圍;當瘤細胞數量較多,所占比例較大時,粒細胞系、紅細胞系及巨核細胞均可明顯減少。值得提出的是,在部分患者,特別在病程早期,骨髓瘤細胞可呈灶性分布,單個部位骨髓穿刺不一定檢出骨髓瘤細胞,此時應作多部位骨髓穿刺或骨髓活檢,方可發現瘤細胞。瘤細胞易位於塗片尾部,應注意檢查塗片尾部。骨髓瘤細胞形態呈多樣性。分化良好者與正常成熟漿細胞形態相似,分化不良者呈典型骨髓瘤細胞形態,而多數瘤細胞形態似幼漿細胞或漿母細胞形態。同一患者的骨髓中可出現形態不一的骨髓瘤細胞。典型骨髓瘤細胞較成熟漿細胞大,直徑為30~50μm細胞外形不規則,可有偽足,胞質藍染,核旁空暈消失或不明顯,胞質中可見泡壁含核糖核酸、泡內含中性核蛋白的空泡,也可見到含本-周蛋白的類棒狀小體,以及外層含免疫球蛋白,而內含糖蛋白的拉塞爾小體(Ruseu小體),核較大,核染色質細緻,有一或兩個核仁。少數瘤細胞具有雙核或多核,但核分裂並不常見。IgA型骨髓瘤細胞胞質經瑞特染色可呈火焰狀,此因嗜鹼性糖蛋白被嗜酸性糖蛋白取代的緣故。據觀察,瘤細胞形態近似成熟漿細胞者病程進展較慢,瘤細胞形態呈分化不良者病程進展較快。在透射電子顯微鏡下,瘤細胞的顯著特徵是內質網的增多和擴大,高爾基(Golgi)體極為發達。擴大的粗面內質網內含無定形物、橢圓形小體,這些物質與血清中M蛋白有關。發達的高爾基體內含緻密小體和空泡。線粒體也增多、增大,嵴豐富。常可見到胞質內有空泡、拉塞爾小體、結晶體、包涵體。胞核大而圓,常偏於一側,核染色質較粗,核仁大而多形化,有時可見核內包涵體。胞核與胞質發育成熟程度不成比例是瘤細胞在透射電子顯微鏡下的重要特徵。應用抗免疫球蛋白的重鏈抗體和抗免疫球蛋白輕鏈抗體,進行免疫熒光法檢查,可發現骨髓瘤細胞呈陽性,但僅含有一種重鏈和一種輕鏈,與其血清中M蛋白(M protein)的重鏈、輕鏈類型一致。
- 血清異常單克隆免疫球蛋白:異常單克隆免疫球蛋白增多引起的高球蛋白血症是本病的重要特徵之一。血清清蛋白減少或正常,A/G比例常倒置。異常單克隆免疫球蛋白大量增多的同時,正常免疫球蛋白常明顯減少。檢測血清異常單克隆免疫球蛋白的方法有下述幾種:
- 血清蛋白醋酸纖維薄膜電泳:異常增多的單克隆免疫球蛋白表現為一濃集的窄帶,經密度掃描儀繪出的圖像表現為一窄底高峰,其峰高度至少較峰底寬度大2倍以上,即M成分(或稱M蛋白)。這是由於單克隆免疫球蛋白的相對分子質量大小、氨基酸組成、所帶電荷完全相同,因而在電場的泳動速度完全相同的緣故。M成分可出現在γ區(IgG,IgM)、β或α2區(IgA),這取決於單克隆免疫球蛋白的類型。當M成分顯著增多時,其他免疫球蛋白及血清清蛋白常明顯減少。
- 免疫電泳:單克隆免疫球蛋白在免疫電泳上表現為異常沉澱弧,在出現一種異常重鏈沉澱弧和一種異常輕鏈沉澱弧的同時,另一種輕鏈和其他類型重鏈常明顯減少。根據免疫電泳結果可以確定單克隆免疫球蛋白類型,從而對多發性骨髓瘤進行分型,即IgG型、IgA型、IgM型、IgD型、IgE型、輕鏈型、雙克隆或多克隆型、不分泌型。
- 聚合酶鏈反應(PCR):近年來採用PCR技術檢測免疫球蛋白重鏈基因重排作為單克隆B細胞——漿細胞惡性增生的標記,用於本病的診斷及與良性反應性免疫球蛋白增多的鑑別診斷。用上述方法檢出單克隆免疫球蛋白後,尚需進行定量,目前多採用速率散射比濁法(rate nephelometry)確定免疫球蛋白濃度。
- 尿液:常規檢查常發現有蛋白尿、鏡下血尿,但管型少見,有時可見到漿(瘤)細胞。具有診斷意義的是尿中出現本周蛋白,又稱凝溶蛋白,該蛋白在酸化的尿液中加熱至50~60℃時發生凝固,但進一步加熱則又溶解。本-周蛋白就是自腎臟排出的免疫球蛋白輕鏈。在多發性骨髓瘤,瘤細胞不僅合成和分泌大量單克隆免疫球蛋白,而且重鏈與輕鏈的合成比例失調,往往有過多輕鏈生成,故血中輕鏈濃度明顯升高。輕鏈的相對分子質量僅23000,可通過腎小球基底膜而排出,故出現本-周蛋白尿。由於單克隆漿(瘤)細胞儀能合成一種輕鏈(κ或λ鏈),故本-周蛋白僅為一種輕鏈。應用免疫電泳可確定本-周蛋白為何種輕鏈。近年來採用速率散射比濁法定量測定尿中輕鏈含量,顯著提高了尿液輕鏈檢測的敏感度和精確度。既往用酸加熱法檢測本-周蛋白的陽性率為30%~60%,且有假陽性。而採用尿液輕鏈定量法的陽性率幾近100%,且不出現假陽性。正常人尿中有κ和λ兩種輕鏈,含量均低。尿中出現大量單一輕鏈,而另一種輕鏈含量減低甚至檢測不出,是MM的特徵之一。
- 腎功能:腎功能常受損,尤多見於病程中期、晚期。血肌酐、尿素氮、內生肌酐清除率測定、酚紅排泄試驗、放射性核素腎圖等檢查可確定腎功能是否受損及受損程度。晚期可發生尿毒症,成為死因之一。當患者有大量本-周蛋白尿時,應避免進行靜脈腎盂造影,因造影劑可能與本-周蛋白發生反應而導致急性腎功能衰竭。
- 血液生化異常:血鈣常升高,國外報告高鈣血症在MM的發生率為30%~60%,國內報告發生率為15%
其他輔助檢查
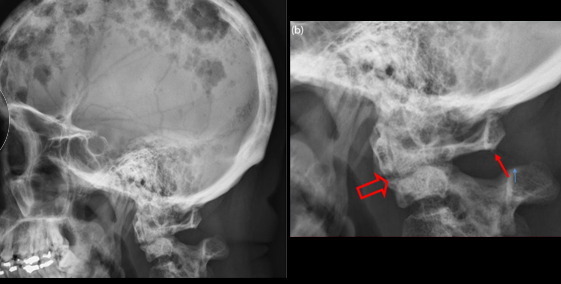
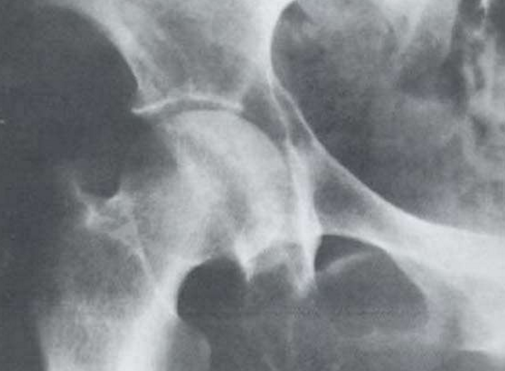
- X射線及其他影像學檢查:X射線檢查在本病診斷上具有重要意義。本病的X射線表現有下述4種:①瀰漫性骨質疏鬆:瘤細胞浸潤及瘤細胞分泌激活破骨細胞的因子(IL-1、淋巴細胞毒素、TNF、OAF)引起普遍性骨質疏鬆。脊椎骨、肋骨、盆骨、顱骨常表現明顯,也可見於四肢長骨。②溶骨性病變:骨質疏鬆病變的進一步發展即造成溶骨性病變。多發性圓形或卵圓形、邊緣清晰銳利似穿鑿樣溶骨性病變是本病的典型X射線徵象,常見於顱骨、盆骨、肋骨、脊椎骨,偶見於四肢骨骼。③病理性骨折:骨折在骨質破壞的基礎上發生,最常見於下胸椎和上腰椎,多表現為壓縮性骨折。其次見於肋骨、鎖骨、盆骨,偶見於四肢骨骼。④骨質硬化:此種病變少見,一般表現為局限性骨質硬化,出現在溶骨性病變周圍。瀰漫性骨質硬化罕見。IgD型骨髓瘤較易並發骨質硬化。γ-骨顯像是近年來用於檢查骨質異常的手段之一。在本病,溶骨性病變表現為病變部位有放射線濃集。此法可一次顯示周身骨骼,且較X射線敏感。X射線僅在骨骼脫鈣達30%以上時才能顯示出病變,而γ-骨顯像在病變早期即可出現放射線濃集徵象。但值得指出的是,γ-骨顯像雖然敏感性較高,但特異性卻不高,任何原因引起的骨質代謝增高均可導致放射線濃集徵象,故應注意鑑別。CT和磁共振成像(MRI)也用於本病的診斷性檢查,特別當骨髓瘤侵犯中樞神經系統或脊椎骨壓縮性骨折損傷脊髓、神經根時,CT及(或)MRI檢查可為診斷提供重要信息。
- B超:腎功能損害,泌尿結石、心肌肥厚者可提示。
- 放射性核素:腎圖檢查可確定腎功能損害程度。
發病機制

關於骨髓瘤細胞的起源,最初依據細胞形態及分泌免疫球蛋白的特點,認為源於漿細胞的惡變。爾後的免疫學和分子生物學研究提示骨髓瘤細胞起始於早期前B細胞(pre-B cell)惡變,其根據是MM患者除有單克隆惡變漿細胞外,尚有單克隆淋巴細胞,該淋巴細胞表面的免疫球蛋白及免疫球蛋白基因重排與瘤細胞相同,早期前B細胞胞質IgM可與抗M蛋白抗體發生特異結合反應。但是,近年來的研究又發現骨髓瘤細胞不僅具有漿細胞和B細胞特徵,而且還表達髓系細胞、紅系細胞、巨核細胞及T細胞表面抗原。還有研究提示T細胞和B細胞的共同前體細胞發生了與瘤細胞相同的免疫球蛋白基因重排,某些MM患者的T細胞亞群能和M蛋白發生特異交叉反應。基於上述研究發現,目前認為MM瘤細胞雖然主要表達B細胞——漿細胞特點,但其起源卻是較前B細胞更早的造血前體細胞(hematopoiesis precursor cell)的惡變。
至於造血前體細胞發生惡變的機制,目前尚未完全闡明。有眾多證據表明MM的發生與癌基因有關。對誘導產生的小鼠漿細胞瘤的研究發現,90%鼠發生染色體易位,而斷裂點幾乎都出現在癌基因C-MYC區,形成重組C-MYC(rC-MYC)並得到表達,提示鼠漿細胞瘤與C-MYC有關。在MM患者中已發現有C-MYC基因重排、突變及mRNA水平升高。癌基因N-RAS或K-RAS突變見於27%(18%~47%)初診MM病例及46%(35%~71%)治療後MM病例。N-RAS突變可導致瘤細胞缺失IL-6條件下,被其他造血因子激活而增殖並減少凋亡。P21的高水平見於部分MM患者,P2l是癌基因H-RAS的產物,表明部分MM患者有癌基因H-RAS的高表達。在動物試驗中,將點突變激活的H-RAS基因植入經EB病毒感染的人B細胞,結果導致B細胞轉化為惡性漿細胞,表現出能在半固體培養基上生長,以及使裸鼠生長腫瘤並分泌大量IgM等惡性漿細胞特徵。
對MM的染色體研究,雖未發現具有標記性的染色體異常,但已肯定出現在MM的一些染色體異常並非是隨機性的,其中1,14號染色體重排最為常見。其次3,5,7,9,11號染色體的三體性和8,13號染色體的單體性,以及6號染色體長臂缺失。也較多見於MM。已有研究證明6號染色體長臂缺失與破骨細胞激活因子(osteoclast activating factor OAF)及腫瘤壞死因子(TNF)生成增多有關,7號染色體異常與多藥耐藥基因(MDR1)表達有關,8號染色體異常與C-MYC癌基因激活有關。因此,目前一般認為,放射線、化學物質、病毒感染等因素可能引起基因突變或染色體易位,激活癌基因,如點突變激活H-RAS和基因重排,激活C-MYC,導致腫瘤發生。關於染色體異常與癌基因的激活,以及癌基因激活與MM發病之間關係的研究目前正在深入研究之中。
淋巴因子細胞因子、生長因子、白細胞介素、集落刺激因子與骨髓瘤的關係在近年來受到重視。B細胞的增生、分化、成熟至漿細胞的過程與多種淋巴因子有關:白細胞介素-1(IL-1)可激活IL-2基因表達;IL-2和IL-3促使早期B細胞增生、分化;IL-4可以激活休止期B細胞,促進B細胞增生;IL-5促使B細胞進一步增生、分化;IL-6刺激B細胞增生並最終分化為產生免疫球蛋白的漿細胞;IL-10可促進B細胞向漿細胞分化並直接刺激骨髓瘤細胞增生,但IL-10水平在MM中很低而在漿細胞白血病中顯著升高,故推測IL-10與MM的晚期病變有關。其中IL-6受到特別注意,因為無論在體內還是在體外,IL-6均可促使漿細胞和骨髓瘤細胞增生,而處於進展期的多發性骨髓瘤患者體內,尤其是骨髓中IL-6水平顯著高於正常。
有實驗證明IL-6可促進BCL-XL表達,抑制瘤細胞凋亡。但是對於IL-6是來自正常組織的旁分泌還是骨髓瘤細胞的自分泌,尚存在着不同意見。有些研究者根據人骨髓瘤細胞株RPMI 8226和U266不分泌IL-6這一現象,提出升高的IL-6可能來自骨髓中單核細胞和間質細胞的旁分泌,而非瘤細胞的自分泌。然而多數研究者認為,儘管單核細胞、骨髓間質細胞、T細胞、內皮細胞、腎小球細胞、角化細胞均可分泌IL-6,但骨髓瘤細胞(包括不同株的RPMI 8226和U266)也可自行分泌IL-6。C反應蛋白(CRP)的水平受IL-6的調節,當IL-6水平升高時,CRP水平也隨之升高,故CRP水平可間接反映IL-6水平。MM患者的CRP水平常升高。根據多種淋巴因子,尤其是IL-6,是B細胞——漿細胞的生長因子和分化因子,進展性






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
用藥治療
(1)陰虛夾瘀:
- 治法:滋補肝腎,活血化瘀。
- 方藥:杞菊地黃丸合桃紅四物湯加減。杞菊地黃丸重在滋陰補腎、清利頭目。方中熟地、山萸肉滋腎填精,山藥補脾固精,茯苓淡滲脾濕,丹皮清瀉虛火,澤瀉通調水道,枸芪子、菊花清利頭目。桃紅四物湯為活血輕劑,方中桃仁、紅花、當歸、赤芍、川芎、熟地具有活血養血之功效。
- 若腎陰虛明顯可酌加女貞子、旱蓮草;無浮腫者可減澤瀉、茯苓;骨痛劇者可適當加用乳香、沒藥。
(2)陽虛痰阻:
- 治法:溫補脾腎,化痰通絡。
- 方藥:陽合湯和消瘰丸加減。陽和湯中的熟地滋腎填精,鹿角膠補腎壯陽;肉桂、姜炭溫陽散寒以通滯;白芥子祛出皮里膜外之痰;麻黃開發腠理,引導寒毒之邪從外解;生甘草解毒。消瘰丸中的玄參、貝母、牡蠣化痰散結。可適當加入昆布、夏枯草、南星、山慈菇等以增強散結之功效;浮腫可加豬苓、茯苓利水消腫。
(3)氣血兩虛:
- 治法:滋腎填精,益氣養血。
- 方藥:六味地黃丸合當歸補血湯加減。六味地黃丸中的熟地、山萸肉滋腎填精;山藥補脾固精;茯苓健脾祛濕;丹皮清瀉虛火;澤瀉通調水道;黃芪、當歸益氣生血。方中可適當加用太子參、黃精、首烏等益氣生血之品。
(4)熱毒熾盛:
飲食保健
- 飲食宜清淡,選用抑制骨髓過度增生的食品,如海帶、紫菜、[裙帶萊]]、海蛤、杏仁。對症選用抗血栓、補血、壯骨和減輕脾腫大的食品,穩定期可長期服用"康血寧"藥茶。
- 抑制骨髓過度增生食療方 桃花魚片:青魚肉適量,桃仁酥10g。魚肉切絲,共炒熟即可。適用於各型多發性骨髓瘤。
- 抗血栓食療方 山楂甜羹:山楂50g,紅花50g。煮羹作點心食。適用於伴有高粘滯血症的多發性骨髓瘤患者。
- 補血、抗消耗食療方
黃芪銀耳湯:黃芪9g,銀耳12g,加水300ml,文火煮1小時加冰糖適量,每日服一次。治療多發性骨髓瘤緩解期,氣陰虛,口乾,盜汗,失眠者。
以上資料僅供參考,詳情請詢醫生。
預防護理
避免或減少有害物質的接觸。
預後
與本病預後有關的因素有:臨床分期(包括腎功能)、免疫球蛋白分型、漿(瘤)細胞分化程度、血清β2-微球蛋白水平、血清乳酸脫氫酶水平以及漿細胞標記指數。臨床分期IA的中數生存期可達5年,而臨床分期ⅢB的中數生存期則短於2年。免疫球蛋白類型對預後也有影響。輕鏈型預後較差,IgA型預後也遜於IgG型。漿(瘤)細胞分化不良者預後劣於漿(瘤)細胞分化較好者。β2-微球蛋白(β2-microglobulin,β2-M)系低相對分子質量(11800)蛋白,是HLA-A、B、C組織相容性抗原複合物的輕鏈部分,正常血清β2-M含量<2.7mg/L,幾乎全部由腎臟排出,近端腎小管以胞飲形式攝取,在腎小管細胞溶酶體降解為氨基酸。在本病由於瘤細胞增生、細胞周轉加速及腎功能損害而導致血、尿β2-M水平升高。
目前公認β2-M是本病的重要預後因素,血清β2-M明顯升高為高危因素之一。血清乳酸脫氫酶(LDH)水平升高由組織壞死釋放引起,見於多種炎症、組織或腫瘤壞死,雖不具特異性,但LDH明顯升高是本病的另一高危因素。漿細胞標記指數(plasma cell labelling index,PCL1)代表漿(瘤)細胞合成DNA狀況,反映骨髓瘤進展狀態,PCLI<1.0屬低危組,PCLI<3.0表示骨髓瘤處進展狀態,屬高危組。此外,對於C反應蛋白(CRP)和胸苷激酶(thymidine kinase,TK)是否為獨立的具有預後意義的因素,目前存在不同意見,尚無定論。
本病的病程在不同患者之間有很大差異。按上述預後因素分析,可將本病患者分為低危組、中危組和高危組。目前尚無公認的、統一的劃分標準,下述劃分標準僅作為參考。低危組在診斷時臨床分期為Ⅰ期,如β2-M<2.7mg/L、PCLI<1%,此組中數生存期>5年;中危組在診斷時臨床分期為Ⅱ期,β2-M≥2.7mg/L或PCLI≥1%,此組中數生存期約為3年;高危組在診斷時臨床分期為Ⅲ期,β2-M≥2.7mg/L,同時PCLI≥1%,此組中數生存期約為1年半。就本病總體而言,在目前的以化療為主要治療的條件下,本病患者的中數生存期為30~36個月。導致患者死亡的主要原因是感染、腎功能衰竭、骨髓瘤進展所致周身衰竭或多器官衰竭,少數患者因胃腸道或顱內出血而死亡。約有5%患者轉變為急性白血病,多為急性漿細胞白血病,但也可為急性單核細胞白血病、急性粒-單核細胞白細胞或急性粒細胞白血病。
視頻
視頻